 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1jm0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF FOUR-HELIX BUNDLE MODEL | |||||||||
 要素 要素 | PROTEIN (FOUR-HELIX BUNDLE MODEL) | |||||||||
 キーワード キーワード | DE NOVO PROTEIN / ALPHA-HELICAL BUNDLE / PROTEIN DESIGN | |||||||||
| 機能・相同性 | Immunoglobulin FC, subunit C / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Up-down Bundle / Mainly Alpha / :  機能・相同性情報 機能・相同性情報 | |||||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å | |||||||||
 データ登録者 データ登録者 | Di Costanzo, L. / Geremia, S. | |||||||||
 引用 引用 | ジャーナル: J.Am.Chem.Soc. / 年: 2001 タイトル: Toward the de novo design of a catalytically active helix bundle: a substrate-accessible carboxylate-bridged dinuclear metal center. 著者: Di Costanzo, L. / Wade, H. / Geremia, S. / Randaccio, L. / Pavone, V. / DeGrado, W.F. / Lombardi, A. #1: ジャーナル: Proc.Natl.Acad.Sci.USA / 年: 2000タイトル: Retrostructural Analysis of Metalloproteins: Application to the Design of a Minimal Model for Diiron Proteins 著者: Lombardi, A. / Summa, C.M. / Geremia, S. / Randaccio, L. / Pavone, V. / DeGrado, W.F. #2: ジャーナル: Curr.Opin.Struct.Biol. / 年: 1999タイトル: Tertiary Templates for the Design of Diiron Proteins 著者: Summa, C.M. / Lombardi, A. / Lewis, M. / DeGrado, W.F. #3: ジャーナル: Annu.Rev.Biochem. / 年: 1999タイトル: De Novo Design and Structural Characterization of Proteins and Metalloproteins 著者: DeGrado, W.F. / Summa, C.M. / Pavone, V. / Nastri, F. / Lombardi, A. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1jm0.cif.gz | 81.6 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1jm0.ent.gz | 62.4 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1jm0.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1jm0_validation.pdf.gz | 408.5 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1jm0_full_validation.pdf.gz | 416 KB | 表示 | |
| XML形式データ | 1jm0_validation.xml.gz | 7.5 KB | 表示 | |
| CIF形式データ | 1jm0_validation.cif.gz | 12.6 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/jm/1jm0ftp://data.pdbj.org/pub/pdb/validation_reports/jm/1jm0 | HTTPS FTP |
-関連構造データ

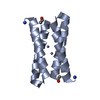








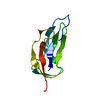

-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 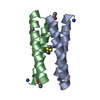
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質・ペプチド | 分子量: 5828.813 Da / 分子数: 6 / 由来タイプ: 合成 / 詳細: THIS PROTEIN WAS CHEMICALLY SYNTHESIZED #2: 化合物 | ChemComp-MN /   分子量: 54.938 Da / 分子数: 11 / 由来タイプ: 合成 / 式: Mn 分子量: 54.938 Da / 分子数: 11 / 由来タイプ: 合成 / 式: Mn#3: 化合物 |   分子量: 78.133 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C2H6OS / コメント: DMSO, 沈殿剤*YM 分子量: 78.133 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C2H6OS / コメント: DMSO, 沈殿剤*YM#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 247 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 247 / 由来タイプ: 天然 / 式: H2OHas protein modification | Y | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.38 Å3/Da / 溶媒含有率: 42 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 277 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7.5 詳細: PEG 400 , Mn(CH3COO)2 , DMSO, Tris-HCl, pH 7.50, VAPOR DIFFUSION, HANGING DROP, temperature 277K | |||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS pH: 7.5 | |||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ELETTRA  / ビームライン: 5.2R / 波長: 1.2 / ビームライン: 5.2R / 波長: 1.2 |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 2000年5月1日 / 詳細: MIRROR |
| 放射 | モノクロメーター: SI(111) / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.2 Å / 相対比: 1 |
| 反射 | 解像度: 1.7→20.1 Å / Num. all: 177455 / Num. obs: 33538 / % possible obs: 99.4 % / Observed criterion σ(F): 0 / 冗長度: 5.3 % / Biso Wilson estimate: 20 Å2 / Rmerge(I) obs: 0.096 / Rsym value: 0.096 / Net I/σ(I): 14 |
| 反射 シェル | 解像度: 1.7→1.8 Å / 冗長度: 5.1 % / Rmerge(I) obs: 0.409 / Mean I/σ(I) obs: 2.8 / Rsym value: 0.409 / % possible all: 96.6 |
| 反射 | *PLUS Num. measured all: 177455 |
| 反射 シェル | *PLUS % possible obs: 96.6 % / Mean I/σ(I) obs: 2.9 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: THEORETICAL MODEL 解像度: 1.7→20.1 Å / Isotropic thermal model: Isotropic / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 26.02 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→20.1 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: REFMAC / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最低解像度: 193.8 Å / Num. reflection obs: 31811 / Rfactor obs: 0.197 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS |
