 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jkq: Testing the Water-Mediated HIN Recombinase DNA Recognition by Sys... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jkq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Testing the Water-Mediated HIN Recombinase DNA Recognition by Systematic Mutations | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA BINDING PROTEIN/DNA / WATER-MEDIATED RECOGNITION / PROTEIN-DNA COMPLEX / HIN RECOMBINASE / G9T MUTANT / DNA BINDING PROTEIN-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA strand exchange activity / DNA integration / DNA recombination / DNA binding Similarity search - Function | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.86 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.86 Å | ||||||
 Authors Authors | Chiu, T.K. / Sohn, C. / Johnson, R.C. / Dickerson, R.E. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 2002 Title: Testing water-mediated DNA recognition by the Hin recombinase. Authors: Chiu, T.K. / Sohn, C. / Dickerson, R.E. / Johnson, R.C. #1: Journal: Thesis / Year: 2001Title: How Hin Recombinase, FIS and Cations Bind DNA. Chapter 4. Water-Mediated Sequence-Specific Recognition by Hin Recombinase Authors: Chiu, T.K. #2: Journal: Science / Year: 1994Title: Hin Recombinase Bound to DNA: The Origin of Specificity in Major and Minor Groove Interactions Authors: Feng, J.A. / Johnson, R.C. / Dickerson, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jkq.cif.gz | 38 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jkq.ent.gz | 24.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1jkq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jk/1jkqftp://data.pdbj.org/pub/pdb/validation_reports/jk/1jkq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ijwSC  1jj6C  1jj8C  1jkoC  1jkpC  1jkrC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

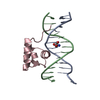

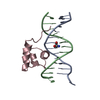







-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 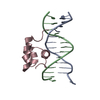
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 4299.824 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: DNA chain | Mass: 4255.831 Da / Num. of mol.: 1 / Source method: obtained synthetically |
| #3: Protein | Mass: 6047.051 Da / Num. of mol.: 1 / Fragment: RESIDUES 139 TO 190 / Source method: obtained synthetically / Details: SYNTHETIC PEPTIDE / References: UniProt: P03013 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 53.36 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 8.5 Details: HANGING DROP VAPOR DIFFUSION AT 4C, WITH INITIAL CONCENTRATION IN DROP OF 0.10 MM DNA, 0.06 MM HIN, 20 MM HEPES (PH 7.5), 20 MM CACL2, 33 MM NACL, 5.0% V/V PEG400, AND 1.56 MM NA CACODYLATE. ...Details: HANGING DROP VAPOR DIFFUSION AT 4C, WITH INITIAL CONCENTRATION IN DROP OF 0.10 MM DNA, 0.06 MM HIN, 20 MM HEPES (PH 7.5), 20 MM CACL2, 33 MM NACL, 5.0% V/V PEG400, AND 1.56 MM NA CACODYLATE. RESERVOIR SOLUTION CONTAINS 100 MM HEPES (PH 7.5), 100 MM CACL2, 100 MM NACL, AND 25% PEG400. CONCENTRATION OF PEG400 IN RESERVOIR SOLUTION WAS INCREASED IN 5% INCREMENTS TO 35%., pH 8.50, VAPOR DIFFUSION, HANGING DROP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 8.5 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 / Beamline: X25 / Wavelength: 1.1 |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Sep 1, 1997 |
| Radiation | Protocol: MOLECULAR REPLACEMENT WITH 1IJW HAVING THE PROPER DNA SUBSTITUTIONS AS THE STARTING MODEL. Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.86→41.3 Å / Num. obs: 3502 / % possible obs: 89.63 % / Observed criterion σ(I): 0 / Redundancy: 12 % / Biso Wilson estimate: 44 Å2 / Rsym value: 0.188 / Net I/σ(I): 15.26 |
| Reflection shell | Resolution: 2.86→2.99 Å / Redundancy: 2.04 % / Mean I/σ(I) obs: 3 / Rsym value: 0.188 / % possible all: 80.76 |
| Reflection | *PLUS Rmerge(I) obs: 0.076 |
| Reflection shell | *PLUS % possible obs: 80.8 % / Rmerge(I) obs: 0.188 / Mean I/σ(I) obs: 3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1IJW Resolution: 2.86→41.3 Å / Isotropic thermal model: ANISOTROPIC_FIXED_ISOTROPIC / σ(F): 0 / Stereochemistry target values: MLF
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 100 Å2 / ksol: 0.35 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.86→41.3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.86→2.99 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 10 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
