 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1j53: Structure of the N-terminal Exonuclease Domain of the Epsilon Sub... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1j53 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of the N-terminal Exonuclease Domain of the Epsilon Subunit of E.coli DNA Polymerase III at pH 8.5 | ||||||
 Components Components | DNA polymerase III, epsilon chain | ||||||
 Keywords Keywords | TRANSFERASE / DNA polymerase proofreading domain | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA polymerase III, core complex / DNA replication proofreading / DNA polymerase III complex / lagging strand elongation / replisome / exonuclease activity / leading strand elongation / 3'-5' exonuclease activity / DNA-templated DNA replication / DNA-directed DNA polymerase ...DNA polymerase III, core complex / DNA replication proofreading / DNA polymerase III complex / lagging strand elongation / replisome / exonuclease activity / leading strand elongation / 3'-5' exonuclease activity / DNA-templated DNA replication / DNA-directed DNA polymerase / DNA-directed DNA polymerase activity / DNA binding / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.8 Å | ||||||
 Authors Authors | Hamdan, S. / Carr, P.D. / Brown, S.E. / Ollis, D.L. / Dixon, N.E. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Structural Basis for Proofreading during Replication of the Escherichia coli Chromosome Authors: Hamdan, S. / Carr, P.D. / Brown, S.E. / Ollis, D.L. / Dixon, N.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1j53.cif.gz | 55.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1j53.ent.gz | 39.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1j53.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j5/1j53ftp://data.pdbj.org/pub/pdb/validation_reports/j5/1j53 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 20741.689 Da / Num. of mol.: 1 / Fragment: N-terminal exonuclease domain (residues 1-186) Source method: isolated from a genetically manipulated source Source: (gene. exp.)   Enterobacteria phage T7 (virus) / References: UniProt: P03007, DNA-directed DNA polymerase Enterobacteria phage T7 (virus) / References: UniProt: P03007, DNA-directed DNA polymerase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#3: Chemical | 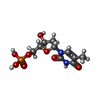  Mass: 322.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N2O8P Mass: 322.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N2O8P#4: Chemical |   Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 229 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 229 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.47 Å3/Da / Density % sol: 50.29 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: PEG 8000, magnesium sulfate, cacodylate, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.5 / Details: Hamdan, S., (2000) J.Struct.Biol., 131, 164. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.9796 / Beamline: BM30A / Wavelength: 0.9796 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Apr 5, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9796 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→50 Å / Num. obs: 20091 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 18.1 % / Rmerge(I) obs: 0.041 / Net I/σ(I): 6.8 |
| Reflection shell | Resolution: 1.8→1.86 Å / Rmerge(I) obs: 0.204 / Num. unique all: 1920 / % possible all: 98.4 |
| Reflection | *PLUS Rmerge(I) obs: 0.041 |
| Reflection shell | *PLUS Rmerge(I) obs: 0.204 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.8→50 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: maximum likelihood target refinements (positional, individual B-factor, and simulated annealing using standard CNS scripts). The TMP 2100 molecule was fitted into the electron density but ...Details: maximum likelihood target refinements (positional, individual B-factor, and simulated annealing using standard CNS scripts). The TMP 2100 molecule was fitted into the electron density but not refined (as attempts to do so increased the Rfree value). The occupancies are zero.
| ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→50 Å
| ||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.227 / Rfactor Rwork: 0.199 | ||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||
| Displacement parameters | *PLUS |
