 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ilz: OUTER MEMBRANE PHOSPHOLIPASE A FROM ESCHERICHIA COLI N156A ACTIVE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ilz | ||||||
|---|---|---|---|---|---|---|---|
| Title | OUTER MEMBRANE PHOSPHOLIPASE A FROM ESCHERICHIA COLI N156A ACTIVE SITE MUTANT pH 6.1 | ||||||
 Components Components | OUTER MEMBRANE PHOSPHOLIPASE A | ||||||
 Keywords Keywords | HYDROLASE / MEMBRANE PROTEIN / ANTI-PARALLEL BETA BARREL / MEMBRANE PHOSPHOLIPASE / SERINE HYDROLASE / CATALYTIC TRIAD / ASN ALA MUTATION | ||||||
| Function / homology |  Function and homology information Function and homology informationphospholipase A1 / glycerophospholipase activity / glycerophospholipid phospholipase A1 activity / phosphatidylglycerol metabolic process / A2-type glycerophospholipase activity / phospholipase A2 / phosphatidylcholine lysophospholipase A1 activity / lipid catabolic process / cell outer membrane / calcium ion binding / protein homodimerization activity Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Snijder, H.J. / Van Eerde, J.H. / Kingma, R.L. / Kalk, K.H. / Dekker, N. / Egmond, M.R. / Dijkstra, B.W. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2001 Title: Structural investigations of the active-site mutant Asn156Ala of outer membrane phospholipase A: function of the Asn-His interaction in the catalytic triad. Authors: Snijder, H.J. / Van Eerde, J.H. / Kingma, R.L. / Kalk, K.H. / Dekker, N. / Egmond, M.R. / Dijkstra, B.W. #1: Journal: Nature / Year: 1999Title: Structural Evidence for Dimerization-Regulated Activation of an Integral Membrane Phospholipase Authors: Snijder, H.J. / Ubarretxena-Belandia, I. / Blaauw, M. / Kalk, K.H. / Verheij, H.M. / Egmond, M.R. / Dekker, N. / Dijkstra, B.W. #2: Journal: FEBS Lett. / Year: 1995Title: Crystallization and Preliminary X-Ray Analysis of Outer Membrane Phospholipase A from Escherichia Coli Authors: Blaauw, M. / Dekker, N. / Verheij, H.M. / Kalk, K.H. / Dijkstra, B.W. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN ATOMS C5'-C8' OF BOG 503 LACK INTERPRETABLE ELECTRON DENSITY. ATOMS C3'-C8' OF BOG 504 ...HETEROGEN ATOMS C5'-C8' OF BOG 503 LACK INTERPRETABLE ELECTRON DENSITY. ATOMS C3'-C8' OF BOG 504 HAVE MISSING ELECTRON DENSITY. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ilz.cif.gz | 69.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ilz.ent.gz | 50.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1ilz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/il/1ilzftp://data.pdbj.org/pub/pdb/validation_reports/il/1ilz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ildC  1im0C  1qd5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31493.852 Da / Num. of mol.: 1 / Mutation: N156A Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||
|---|---|---|---|---|---|
| #2: Sugar | ChemComp-BOG / 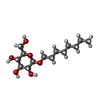  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 5 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 5Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM #3: Chemical |   Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 32 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.79 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.1 Details: MPD, calcium chloride, bis-tris buffer, pH 6.1, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.6 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ELETTRA  / Beamline: 5.2R / Wavelength: 1 Å / Beamline: 5.2R / Wavelength: 1 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→31 Å / Num. all: 12728 / Num. obs: 150605 / % possible obs: 98.8 % / Observed criterion σ(I): -3 / Redundancy: 11.8 % / Rmerge(I) obs: 0.037 / Rsym value: 0.037 / Net I/σ(I): 39.2 |
| Reflection shell | Resolution: 2.5→2.54 Å / Rmerge(I) obs: 0.113 / Mean I/σ(I) obs: 8.3 / Num. unique all: 611 / Rsym value: 0.113 / % possible all: 96.1 |
| Reflection | *PLUS Lowest resolution: 31 Å / Num. obs: 12728 / Num. measured all: 150605 |
| Reflection shell | *PLUS % possible obs: 96.1 % / Num. unique obs: 611 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: Starting model 1QD5 with the active site residues truncated to ala and all waters and detergent molecules removed. All remaining atoms have been given a random shift of almost 0.5 ...Starting model: Starting model 1QD5 with the active site residues truncated to ala and all waters and detergent molecules removed. All remaining atoms have been given a random shift of almost 0.5 Angstrom in all directions. Resolution: 2.5→31 Å / Isotropic thermal model: anisotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): -3 / Stereochemistry target values: Engh & Huber Details: Bulk solvent correction was applied as implemented in CNS bulk solvent: density level= 0.379342 e/A^3, B-factor= 52.8672 A^2
| |||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→31 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 31 Å / σ(F): 0 / % reflection Rfree: 9.7 % / Rfactor obs: 0.222 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
