 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1igb: AEROMONAS PROTEOLYTICA AMINOPEPTIDASE COMPLEXED WITH THE INHIBITO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1igb | ||||||
|---|---|---|---|---|---|---|---|
| Title | AEROMONAS PROTEOLYTICA AMINOPEPTIDASE COMPLEXED WITH THE INHIBITOR PARA-IODO-D-PHENYLALANINE HYDROXAMATE | ||||||
 Components Components | AMINOPEPTIDASE | ||||||
 Keywords Keywords | AMINOPEPTIDASE / HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationbacterial leucyl aminopeptidase / metalloexopeptidase activity / aminopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  Vibrio proteolyticus (bacteria) Vibrio proteolyticus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) / Resolution: 2.3 Å | ||||||
 Authors Authors | Chevrier, B. / D'Orchymont, H. / Schalk, C. / Tarnus, C. / Moras, D. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1996 Title: The structure of the Aeromonas proteolytica aminopeptidase complexed with a hydroxamate inhibitor. Involvement in catalysis of Glu151 and two zinc ions of the co-catalytic unit. Authors: Chevrier, B. / D'Orchymont, H. / Schalk, C. / Tarnus, C. / Moras, D. #1: Journal: Structure / Year: 1994Title: Crystal Structure of Aeromonas Proteolytica Aminopeptidase: A Prototypical Member of the Co-Catalytic Zinc Enzyme Family Authors: Chevrier, B. / Schalk, C. / D'Orchymont, H. / Rondeau, J.M. / Moras, D. / Tarnus, C. #2: Journal: Arch.Biochem.Biophys. / Year: 1992Title: Rapid Purification of the Aeromonas Proteolytica Aminopeptidase: Crystallization and Preliminary X-Ray Data Authors: Schalk, C. / Remy, J.M. / Chevrier, B. / Moras, D. / Tarnus, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1igb.cif.gz | 92.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1igb.ent.gz | 70.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1igb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ig/1igbftp://data.pdbj.org/pub/pdb/validation_reports/ig/1igb | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31427.350 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Vibrio proteolyticus (bacteria)References: UniProt: Q01693, bacterial leucyl aminopeptidase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-IPO / | 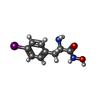  Mass: 306.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11IN2O2 Mass: 306.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11IN2O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 220 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 220 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.69 Å3/Da / Density % sol: 54.27 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 291 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: SIEMENS / Wavelength: 1.5418 |
| Detector | Type: XENTRONICS / Detector: AREA DETECTOR / Date: Sep 1, 1993 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→50 Å / Num. obs: 15274 / % possible obs: 91.7 % / Observed criterion σ(I): 0 / Redundancy: 7.7 % / Rmerge(I) obs: 0.081 |
| Reflection | *PLUS Num. measured all: 118144 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT SOFTWARE USED : X-PLOR STARTING MODEL FOR MOLECULAR REPLACEMENT: NATIVE PROTEIN (REFERENCE 1) Resolution: 2.3→8 Å / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.01 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.16 / Rfactor Rwork: 0.16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
