 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hg5: CALM-N N-terminal domain of clathrin assembly lymphoid myeloid le... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hg5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CALM-N N-terminal domain of clathrin assembly lymphoid myeloid leukaemia protein, inositol(1,2,3,4,5,6)P6 complex | ||||||
 Components Components | CLATHRIN ASSEMBLY PROTEIN SHORT FORM | ||||||
 Keywords Keywords | ENDOCYTOSIS / ADAPTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationRND3 GTPase cycle / vesicle cargo loading / membrane bending / endosome to plasma membrane transport vesicle / regulation of terminal button organization / 1-phosphatidylinositol binding / positive regulation of synaptic vesicle clustering / regulation of protein transport / extrinsic component of presynaptic endocytic zone membrane / amyloid-beta clearance by transcytosis ...RND3 GTPase cycle / vesicle cargo loading / membrane bending / endosome to plasma membrane transport vesicle / regulation of terminal button organization / 1-phosphatidylinositol binding / positive regulation of synaptic vesicle clustering / regulation of protein transport / extrinsic component of presynaptic endocytic zone membrane / amyloid-beta clearance by transcytosis / synaptic vesicle maturation / regulation of synaptic vesicle transport / clathrin coat of coated pit / Golgi Associated Vesicle Biogenesis / negative regulation of protein localization to cell surface / regulation of vesicle size / positive regulation of amyloid precursor protein catabolic process / postsynaptic endocytic zone / clathrin heavy chain binding / positive regulation of dendrite extension / Cargo recognition for clathrin-mediated endocytosis / clathrin coat assembly / Clathrin-mediated endocytosis / positive regulation of synaptic vesicle endocytosis / vesicle budding from membrane / clathrin-dependent endocytosis / positive regulation of Ras protein signal transduction / dendrite morphogenesis / positive regulation of axonogenesis / regulation of amyloid precursor protein catabolic process / clathrin-coated vesicle / endosomal transport / positive regulation of amyloid-beta formation / low-density lipoprotein particle receptor binding / neurofibrillary tangle / clathrin binding / parallel fiber to Purkinje cell synapse / regulation of endocytosis / hemopoiesis / regulation of synaptic vesicle endocytosis / negative regulation of protein localization to plasma membrane / synaptic vesicle endocytosis / vesicle-mediated transport / axonogenesis / phosphatidylinositol-4,5-bisphosphate binding / clathrin-coated pit / receptor-mediated endocytosis / SNARE binding / receptor internalization / negative regulation of receptor-mediated endocytosis / SH3 domain binding / multicellular organismal-level iron ion homeostasis / tau protein binding / Schaffer collateral - CA1 synapse / small GTPase binding / endocytosis / regulation of protein localization / synaptic vesicle / presynaptic membrane / vesicle / intracellular iron ion homeostasis / learning or memory / early endosome / postsynaptic membrane / postsynapse / endosome / postsynaptic density / negative regulation of gene expression / neuronal cell body / positive regulation of DNA-templated transcription / perinuclear region of cytoplasm / Golgi apparatus / cell surface / membrane / identical protein binding / nucleus / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Ford, M.G.J. / Evans, P.R. / McMahon, H.T. | ||||||
 Citation Citation | Journal: Science / Year: 2001 Title: Simultaneous Binding of Ptdins(4,5)P2 and Clathrin by Ap180 in the Nucleation of Clathrin Lattices on Membranes Authors: Ford, M.G.J. / Pearse, B.M.F. / Higgins, M.K. / Vallis, Y. / Owen, D.J. / Gibson, A. / Hopkins, C.R. / Evans, P.R. / Mcmahon, H.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hg5.cif.gz | 69.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hg5.ent.gz | 51.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1hg5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hg/1hg5ftp://data.pdbj.org/pub/pdb/validation_reports/hg/1hg5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hf8SC  1hfaC  1hg2C S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32865.789 Da / Num. of mol.: 1 / Fragment: N-TERMINAL DOMAIN RESIDUES 1-289 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-IHP / 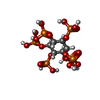  Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 Mass: 660.035 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H18O24P6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 129 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.08 Å3/Da / Density % sol: 60 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 0.1M HEPES, PH 7.5, 12% PEG 8K, 8% ETHYLENE GLYCOL CRYSTALS SOAKED IN 1MM LIGAND FOR 1 HOUR | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: ADSC QUANTUM 4 CCD / Detector: CCD / Date: Mar 3, 2000 |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2→41 Å / Num. obs: 25874 / % possible obs: 100 % / Observed criterion σ(I): 6 / Redundancy: 7 % / Biso Wilson estimate: 43 Å2 / Rmerge(I) obs: 0.082 / Rsym value: 0.082 / Net I/σ(I): 16.3 |
| Reflection shell | Resolution: 2→2.11 Å / Redundancy: 6.4 % / Rmerge(I) obs: 0.952 / Mean I/σ(I) obs: 1.7 / Rsym value: 0.952 / % possible all: 100 |
| Reflection | *PLUS % possible obs: 100 % |
| Reflection shell | *PLUS % possible obs: 100 % / Rmerge(I) obs: 0.00952 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HF8 Resolution: 2→65.94 Å / SU B: 6.90327 / SU ML: 0.18632 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.14963 / ESU R Free: 0.13444 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS SCALING DETAILS BABINET"S PRINCIPLE FOR SCALING HAS BEEN USED BULK SOLVENT CORRECTION BASED ON CONSTANT VALUE HAS BEEN U PARAMETERS FOR MASK ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS SCALING DETAILS BABINET"S PRINCIPLE FOR SCALING HAS BEEN USED BULK SOLVENT CORRECTION BASED ON CONSTANT VALUE HAS BEEN U PARAMETERS FOR MASK CALCULATION VDW PROB RADII = 1.40 ION PROB RADII = 0.80 SHRINKAGE RADII = 0.80
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.384 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→65.94 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.19286 / Rfactor Rfree: 0.21723 / Rfactor Rwork: 0.19159 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 44.384 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
