 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h8p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Bull seminal plasma PDC-109 fibronectin type II module | ||||||
 Components Components | SEMINAL PLASMA PROTEIN PDC-109 | ||||||
 Keywords Keywords | PHOSPHORYLCHOLINE-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of sperm capacitation / phospholipid efflux / sperm capacitation / single fertilization / heparin binding / cell surface / : Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.82 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.82 Å | ||||||
 Authors Authors | Wah, D.A. / Fernandez-Tornero, C. / Calvete, J.J. / Romero, A. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Sperm Coating Mechanism from the 1.8 A Crystal Structure of Pdc-109-Phosphorylcholine Complex Authors: Wah, D.A. / Fernandez-Tornero, C. / Sanz, L. / Romero, A. / Calvete, J.J. #1: Journal: Proteins: Struct.,Funct., Genet. / Year: 1997 Title: Crystallization and Preliminary X-Ray Diffraction Analysis of Bovine Seminal Plasma Pdc-109, a Protein Composed of Two Fibronectin Type II Domains Authors: Romero, A. / Varela, P.F. / Topfer-Petersen, E. / Calvete, J.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h8p.cif.gz | 55.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h8p.ent.gz | 40.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1h8p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h8/1h8pftp://data.pdbj.org/pub/pdb/validation_reports/h8/1h8p | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.93464, -0.35469, -0.02529), Vector: Details | THE PROTEIN IS ACTIVE AS HOMODIMER. | |
-Components
| #1: Protein | Mass: 12811.352 Da / Num. of mol.: 2 / Fragment: RESIDUES 26-134 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-PC / 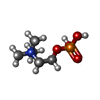  Mass: 184.151 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H15NO4P Mass: 184.151 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C5H15NO4P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | THE SWS ENTRY INCLUDES A PEPTIDE SIGNAL OF 25 AA. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.43 Å3/Da / Density % sol: 49.1 % Description: MAD EXPERIMENT WAS UNDERTAKEN AT DESY-HAMBURG (X31). A NATIVE DATA SET WAS COLLECTED AT ELETTRA (BEAMLINE 5.2 R). | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.2 Details: CRYSTALS WERE OBTAINED IN 30% ISOPROPANOL, 5% PEG 4000, 0.1 M HEPES, PH 7.2. | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 8 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Wavelength: 1.07466,1.07516,0.8856, 1.05271 / Beamline: X31 / Wavelength: 1.07466,1.07516,0.8856, 1.05271 | |||||||||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: May 15, 2000 / Details: TOROIDAL MIRROR | |||||||||||||||
| Radiation | Monochromator: SI(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 1.82→30 Å / Num. obs: 275885 / % possible obs: 99 % / Redundancy: 5.3 % / Biso Wilson estimate: 10.8 Å2 / Rmerge(I) obs: 0.045 / Net I/σ(I): 11.7 | |||||||||||||||
| Reflection shell | Resolution: 1.82→1.92 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.126 / Mean I/σ(I) obs: 5.3 / % possible all: 96.3 | |||||||||||||||
| Reflection | *PLUS Highest resolution: 1.8 Å / Num. obs: 22445 / % possible obs: 99 % / Num. measured all: 275885 / Rmerge(I) obs: 0.032 | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 96.3 % / Rmerge(I) obs: 0.119 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.82→16.32 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1454528.39 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE FIRST 21 RESIDUES (ASP 1-ASP 21) WERE NOT VISIBLE IN THE ELECTRON DENSITY MAPS AT 1.82 A RESOLUTION.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 60.4559 Å2 / ksol: 0.503596 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.82→16.32 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.91 Å / Rfactor Rfree error: 0.016 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 30 Å / Rfactor Rfree: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.202 |
