 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h4g: Oligosaccharide-binding to family 11 xylanases: both covalent int... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h4g | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Oligosaccharide-binding to family 11 xylanases: both covalent intermediate and mutant-product complexes display 2,5B conformations at the active-centre | ||||||||||||
 Components Components | XYLANASE | ||||||||||||
 Keywords Keywords | GLYCOSIDE HYDROLASE / XYLANASE / OLIGOSACCHARIDE / TRANSITION-STATE / INTERMEDIATE / MUTANT / BOAT CONFORMATION | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationendo-1,4-beta-xylanase / endo-1,4-beta-xylanase activity / xylan catabolic process Similarity search - Function | ||||||||||||
| Biological species |  BACILLUS AGARADHAERENS (bacteria) BACILLUS AGARADHAERENS (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.1 Å X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.1 Å | ||||||||||||
 Authors Authors | Sabini, E. / Wilson, K.S. / Danielsen, S. / Schulein, M. / Davies, G.J. | ||||||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Oligosaccharide binding to family 11 xylanases: both covalent intermediate and mutant product complexes display (2,5)B conformations at the active centre. Authors: Sabini, E. / Wilson, K.S. / Danielsen, S. / Schulein, M. / Davies, G.J. #1: Journal: Chem.Biol. / Year: 1999Title: Catalysis and Specificity in Enzymatic Glycoside Hydrolysis: A 2,5B Conformation for the Glycosyl-Enzyme Intermediate Revealed by the Structure of the Bacillus Agaradhaerens Family 11 Xylanase. Authors: Sabini, E. / Sulzenbacher, G. / Dauter, M. / Dauter, Z. / Jorgensen, P.L. / Schulein, M. / Dupont, C. / Davies, G.J. / Wilson, K.S. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h4g.cif.gz | 198.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h4g.ent.gz | 159.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1h4g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h4/1h4gftp://data.pdbj.org/pub/pdb/validation_reports/h4/1h4g | HTTPS FTP |
|---|
-Related structure data
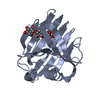



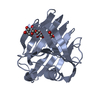




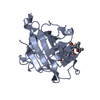

-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23155.547 Da / Num. of mol.: 2 / Fragment: FAMILY 11 XYLANASE CATALYTIC DOMAIN Source method: isolated from a genetically manipulated source Details: 2-FLUORO-2-DEOXY-XYLOBIOSIDE COVALENTLY BOUND TO NUCLEOPHILE GLU94 IN THE ACTIVE SITE Source: (gene. exp.) BACILLUS AGARADHAERENS (bacteria) / Production host: #2: Polysaccharide | Source method: isolated from a genetically manipulated source #3: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 606 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 606 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 53 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 Details: DROP: 2UL PROTEIN (10 MG ML-1 IN 100MM SODIUM ACETATE) PLUS 1UL RESERVOIR RESERVOIR: 100 MM MES PH 6.5, 30% AMMONIUM SULPHATE | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.8469 / Beamline: BW7B / Wavelength: 0.8469 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8469 Å / Relative weight: 1 |
| Reflection | Resolution: 1.1→20 Å / Num. obs: 171783 / % possible obs: 99.8 % / Observed criterion σ(I): 2 / Redundancy: 4.5 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 17.6 |
| Reflection shell | Resolution: 1.1→1.12 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.708 / Mean I/σ(I) obs: 2 / % possible all: 99 |
| Reflection | *PLUS Highest resolution: 1.1 Å / % possible obs: 99.9 % / Rmerge(I) obs: 0.055 |
| Reflection shell | *PLUS % possible obs: 99 % / Rmerge(I) obs: 0.71 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.1→20 Å / SU B: 0.38942 / SU ML: 0.01943 / Cross valid method: THROUGHOUT / ESU R Free: 0.03143
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.1→20 Å
| ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.1 Å / Rfactor obs: 0.158 / Rfactor Rfree: 0.18 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
