 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h1l: NITROGENASE MO-FE PROTEIN FROM KLEBSIELLA PNEUMONIAE, NIFV MUTANT -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h1l | ||||||
|---|---|---|---|---|---|---|---|
| Title | NITROGENASE MO-FE PROTEIN FROM KLEBSIELLA PNEUMONIAE, NIFV MUTANT | ||||||
 Components Components | (NITROGENASE MOLYBDENUM IRON PROTEIN ...) x 2 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / BIOLOGICAL NITROGEN FIXATION / NITROGEN METABOLISM / MOLYBDOENZYMES / ELECTRON TRANSFER | ||||||
| Function / homology |  Function and homology information Function and homology informationmolybdenum-iron nitrogenase complex / nitrogenase / nitrogenase activity / iron-sulfur cluster binding / ATP binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  KLEBSIELLA PNEUMONIAE (bacteria) KLEBSIELLA PNEUMONIAE (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Mayer, S.M. / Gormal, C.A. / Smith, B.E. / Lawson, D.M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Crystallographic Analysis of the Mofe Protein of Nitrogenase from a Nifv Mutant of Klebsiella Pneumoniae Identifies Citrate as a Ligand to the Molybdenum of Iron Molybdenum Cofactor (Femoco). Authors: Mayer, S.M. / Gormal, C.A. / Smith, B.E. / Lawson, D.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h1l.cif.gz | 430.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h1l.ent.gz | 344.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1h1l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h1/1h1lftp://data.pdbj.org/pub/pdb/validation_reports/h1/1h1l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qguS S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj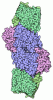
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
-NITROGENASE MOLYBDENUM IRON PROTEIN ... , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 53919.387 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: CHAINS A AND C ARE ALPHA-SUBUNITS / Source: (natural) KLEBSIELLA PNEUMONIAE (bacteria) / Variant: NIFV MUTANT / Strain: UNF1613 / References: UniProt: P00466, nitrogenase#2: Protein | Mass: 58338.180 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Details: CHAINS B AND D ARE BETA-SUBUNITS / Source: (natural) KLEBSIELLA PNEUMONIAE (bacteria) / Variant: NIFV MUTANT / Strain: UNF1613 / References: UniProt: P09772, nitrogenase |
|---|
-Non-polymers , 6 types, 1549 molecules 


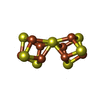


| #3: Chemical |  Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7#4: Chemical |  Mass: 775.440 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe7MoS9 Mass: 775.440 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe7MoS9#5: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#6: Chemical |  Mass: 671.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe8S7 Mass: 671.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe8S7#7: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1537 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | NITROGEN FIXATION, CATALYZED BY THE NITROGENASE COMPLEX, THE IRON PROTEIN AND THE MOLYBDENUM-IRON ...NITROGEN FIXATION, CATALYZED BY THE NITROGENAS |
|---|---|
| Sequence details | N-TERMINAL SEQUENCING INDICATES THAT THE FIRST TWO AMINO ACIDS OF THE ALPHA SUBUNITS (CHAINS A AND ...N-TERMINAL SEQUENCING |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46 % Description: PRIOR TO DATA COLLECTION THE CRYSTAL WAS SOAKED FOR 5 MINS IN CRYOPROTECTANT (MOTHER LIQUOR WITH 25% ETHYLENE GLYCOL CONTAINING 20 MM SODIUM DITHIONITE (A REDUCTANT). | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: liquid diffusion / pH: 7.4 Details: CRYSTALLIZED ANAEROBICALLY USING LIQUID-LIQUID DIFFUSION TECHNIQUE. FINAL CONCENTRATIONS AFTER EQUILIBRATION WERE: 7% (W/V) PEG6000, 0.4 - 0.6 M MGCL2, 50 MM TRIS-HCL PH 7.4, 1.3 MG/ML PROTEIN. | ||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: liquid-liquid diffusion | ||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.936 / Beamline: ID14-4 / Wavelength: 0.936 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Feb 8, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.936 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→40 Å / Num. obs: 154203 / % possible obs: 94.2 % / Observed criterion σ(I): -3 / Redundancy: 3.9 % / Biso Wilson estimate: 20.3 Å2 / Rmerge(I) obs: 0.094 / Net I/σ(I): 15.5 |
| Reflection shell | Resolution: 1.9→1.93 Å / Rmerge(I) obs: 0.214 / Mean I/σ(I) obs: 3.6 / % possible all: 85.2 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 40 Å |
| Reflection shell | *PLUS % possible obs: 85.2 % / Mean I/σ(I) obs: 3.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QGU Resolution: 1.9→40 Å / Cross valid method: THROUGHOUT / σ(F): 0 Details: TOWARDS THE END OF REFINEMENT ALL RESTRAIN WITHIN THE HETEROGENS CIT, CFM, AND CLF WERE EXPLICITL TURNED OFF. IN ADDITION, THE BONDS BETWEEN THESE HETEROGENS AND THE PROTEIN NOT RESTRAINED. ...Details: TOWARDS THE END OF REFINEMENT ALL RESTRAIN WITHIN THE HETEROGENS CIT, CFM, AND CLF WERE EXPLICITL TURNED OFF. IN ADDITION, THE BONDS BETWEEN THESE HETEROGENS AND THE PROTEIN NOT RESTRAINED. BOTH CITRATE MOLECULES WERE REFINED WITH AN OCCUPANCY OF 0.5. A NETWORK OF WATER MOLECULES WAS THOU TO BE PRESENT IN ITS ABSENCE. THREE WATERS WERE MODELLED IN PLA EACH CITRATE AND GIVEN OCCUPANCIES OF 0.5.
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 24 Å2 | ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→40 Å
| ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
