SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED.
Mass: 18.015 Da / Num. of mol.: 173 / Source method: isolated from a natural source / Formula: H2O
Compound details
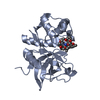
CHAIN A ENGINEERED MUTATION CYS139SER
Has protein modification
Y
Sequence details
REFERENCE: WIEDERANDERS B, ET AL., (1992) J. BIOL. CHEM. 267 PP 13708-13713. THE SEQUENCE USED IS ...REFERENCE: WIEDERANDERS B, ET AL., (1992) J. BIOL. CHEM. 267 PP 13708-13713. THE SEQUENCE USED IS THAT PUBLISHED IN THE ABOVE REFERENCE THIS ACCOUNTS FOR THE TWO CONFLICTS NOTED IN THE SEQADV RECORDS
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.3 Å3/Da / Density % sol: 46 %
Crystal grow
Method: vapor diffusion, hanging drop / pH: 4.1 Details: CRYSTALLISATION WAS PERFORMED BY THE HANGING-DROP VAPOUR DIFFUSION METHOD. EQUAL VOLUMES CATHEPSIN S (CYS25SER) AT 7 MG/ML, INCLUDING THE PEPTIDE ABZ-LEU-THR-BAL-HYP-TYR(NO2)-ASP-NH2 AT 1 MM ...Details: CRYSTALLISATION WAS PERFORMED BY THE HANGING-DROP VAPOUR DIFFUSION METHOD. EQUAL VOLUMES CATHEPSIN S (CYS25SER) AT 7 MG/ML, INCLUDING THE PEPTIDE ABZ-LEU-THR-BAL-HYP-TYR(NO2)-ASP-NH2 AT 1 MM AND WELL SOLUTION WERE COMBINED AND PLACED OVER A WELL CONTAINING 20% ISOPROPANOL, 20% PEG2000 AND 0.1M SODIUM CITRATE, PH 4.13
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads




















 PDBj
PDBj











 Assembly
Assembly

 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P25774, cathepsin S
SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P25774, cathepsin S Mass: 18.015 Da / Num. of mol.: 173 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 173 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing