 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gjn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Hydrogen Peroxide Derived Myoglobin Compound II at pH 5.2 | ||||||
 Components Components | MYOGLOBIN | ||||||
 Keywords Keywords | OXYGEN TRANSPORT / REACTION INTERMEDIATE / HAEM / HEME / OXYGEN ACTIVATION / PEROXIDASE / MONOOXYGENASE / FERRYL / HYDROXY RADICAL | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / oxygen transport / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / skeletal muscle contraction / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding ...Oxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / oxygen transport / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / skeletal muscle contraction / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.35 Å | ||||||
 Authors Authors | Hersleth, H.-P. / Dalhus, B. / Gorbitz, C.H. / Andersson, K.K. | ||||||
 Citation Citation | Journal: J.Biol.Inorg.Chem. / Year: 2002 Title: An Iron Hydroxide Moiety in the 1.35 A Resolution Structure of Hydrogen Peroxide Derived Myoglobin Compound II at Ph 5.2 Authors: Hersleth, H.-P. / Dalhus, B. / Gorbitz, C.H. / Andersson, K.K. #1: Journal: Biochim.Biophys.Acta / Year: 1997Title: A Myoglobin Variant with a Polar Substitution in a Conserved Hydrophobic Cluster in the Heme Binding Pocket Authors: Maurus, R. / Overall, C.M. / Bogumil, R. / Luo, Y. / Mauk, A.G. / Smith, M. / Brayer, G.D. #2: Journal: J.Mol.Biol. / Year: 1987 Title: Crystallization and Preliminary Diffraction Data for Horse Heart Metmyoglobin Authors: Sherwood, C. / Mauk, A.G. / Brayer, G.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gjn.cif.gz | 49.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gjn.ent.gz | 33.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1gjn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gj/1gjnftp://data.pdbj.org/pub/pdb/validation_reports/gj/1gjn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1wlaS S: Starting model for refinement |
|---|---|
| Similar structure data |






























-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 16983.514 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: A FE(III)-BONDED HYDROXYL RADICAL / Source: (natural) | ||
|---|---|---|---|
| #2: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 | ||
| #3: Chemical | ChemComp-OH / 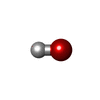  Mass: 17.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO Mass: 17.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO | ||
| #4: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 159 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 159 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 31.76 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.2 Details: 6-10 MG/ML PROTEIN 3.0 M AMMONIUM SULPHATE, 10.8% GLYCEROL PH 5.2, RT. | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.873 / Beamline: BM1A / Wavelength: 0.873 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Feb 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 1.35→21 Å / Num. obs: 26717 / % possible obs: 98.4 % / Redundancy: 4.2 % / Biso Wilson estimate: 11.5 Å2 / Rmerge(I) obs: 0.083 / Net I/σ(I): 22 |
| Reflection shell | Resolution: 1.35→1.38 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.305 / Mean I/σ(I) obs: 2.7 / % possible all: 85.4 |
| Reflection | *PLUS Lowest resolution: 21 Å / Num. measured all: 296944 |
| Reflection shell | *PLUS % possible obs: 85.4 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1WLA Resolution: 1.35→20.79 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 2057058.11 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 71.5094 Å2 / ksol: 0.438879 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.35→20.79 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.35→1.43 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
