 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gi9: A NOVEL SERINE PROTEASE INHIBITION MOTIF INVOLVING A MULTI-CENTER... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gi9 | ||||||
|---|---|---|---|---|---|---|---|
| Title | A NOVEL SERINE PROTEASE INHIBITION MOTIF INVOLVING A MULTI-CENTERED SHORT HYDROGEN BONDING NETWORK AT THE ACTIVE SITE | ||||||
 Components Components | (UROKINASE-TYPE PLASMINOGEN ACTIVATOR) x 2 | ||||||
 Keywords Keywords | BLOOD CLOTTING / hydrolase / three-centered / very short hydrogen bond / oxyanion hole water / shift of pKa of His57 / structure-based drug design / specificity / urokinase / trypsin / thrombin / Zn+2-mediated inhibition | ||||||
| Function / homology |  Function and homology information Function and homology informationu-plasminogen activator / regulation of smooth muscle cell-matrix adhesion / urokinase plasminogen activator signaling pathway / regulation of plasminogen activation / regulation of integrin-mediated signaling pathway / regulation of fibrinolysis / protein complex involved in cell-matrix adhesion / regulation of wound healing / negative regulation of plasminogen activation / serine-type endopeptidase complex ...u-plasminogen activator / regulation of smooth muscle cell-matrix adhesion / urokinase plasminogen activator signaling pathway / regulation of plasminogen activation / regulation of integrin-mediated signaling pathway / regulation of fibrinolysis / protein complex involved in cell-matrix adhesion / regulation of wound healing / negative regulation of plasminogen activation / serine-type endopeptidase complex / regulation of smooth muscle cell migration / Dissolution of Fibrin Clot / regulation of cell adhesion mediated by integrin / smooth muscle cell migration / plasminogen activation / tertiary granule membrane / negative regulation of fibrinolysis / regulation of cell adhesion / serine protease inhibitor complex / specific granule membrane / fibrinolysis / positive regulation of epidermal growth factor receptor signaling pathway / chemotaxis / blood coagulation / regulation of cell population proliferation / response to hypoxia / positive regulation of cell migration / receptor ligand activity / serine-type endopeptidase activity / external side of plasma membrane / focal adhesion / Neutrophil degranulation / cell surface / signal transduction / proteolysis / : / extracellular exosome / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.8 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 1.8 Å | ||||||
 Authors Authors | Katz, B.A. / Elrod, K. / Luong, C. / Rice, M. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hatayte, J. / Janc, J. / Link, J. ...Katz, B.A. / Elrod, K. / Luong, C. / Rice, M. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hatayte, J. / Janc, J. / Link, J. / Litvak, J. / Rai, R. / Rice, K. / Sideris, S. / Verner, E. / Young, W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. Authors: Katz, B.A. / Elrod, K. / Luong, C. / Rice, M.J. / Mackman, R.L. / Sprengeler, P.A. / Spencer, J. / Hataye, J. / Janc, J. / Link, J. / Litvak, J. / Rai, R. / Rice, K. / Sideris, S. / Verner, E. / Young, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gi9.cif.gz | 119.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gi9.ent.gz | 93.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1gi9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gi/1gi9ftp://data.pdbj.org/pub/pdb/validation_reports/gi/1gi9 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ghvC  1ghwC  1ghxC  1ghyC  1ghzC  1gi0C  1gi1C  1gi2C  1gi3C  1gi4C  1gi5C  1gi6C  1gi7C  1gi8C C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 2708.183 Da / Num. of mol.: 1 / Fragment: SHORT CHAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Description: CHEM.BIOL. 7, 299-312, 2000 / Plasmid: PPIC9LMWUPA / Production host:  Pichia pastoris (fungus) / References: UniProt: P00749 Pichia pastoris (fungus) / References: UniProt: P00749 | ||||
|---|---|---|---|---|---|
| #2: Protein | Mass: 27579.473 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN / Mutation: N145A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Description: CHEM.BIOL. 7, 299-312, 2000 / Plasmid: PPIC9LMWUPA / Production host: Pichia pastoris (fungus) / References: UniProt: P00749, u-plasminogen activator | ||||
| #3: Chemical | ChemComp-123 / 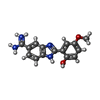  Mass: 283.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H15N4O2 Mass: 283.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H15N4O2 | ||||
| #4: Chemical |   Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H8O7#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 126 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 126 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 40.87 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 6.5 Details: 2-propanol PEG 4000, pH 6.5, vapor diffusion at 298 K, pH 6.50 |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Aug 25, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→38.49 Å / Num. all: 23245 / Num. obs: 22245 / % possible obs: 95.7 % / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Rmerge(I) obs: 0.128 / Net I/σ(I): 5.4 |
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 1.94 % / Num. unique all: 1992 / % possible all: 86.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.8→7 Å / σ(F): 2 / Stereochemistry target values: X-PLOR force field Details: Only Leu_A9 to Thr_A17 are invluded for the A-chain. Residues prior and after these residues are not visible (disordered). Residues after Thr_B242 are not visible (disordered). Residues ...Details: Only Leu_A9 to Thr_A17 are invluded for the A-chain. Residues prior and after these residues are not visible (disordered). Residues after Thr_B242 are not visible (disordered). Residues simultaneously refined in two or more conformations are: Ile_B17, Met_B47, Met_B81, Ser_B95, Arg_B166. No energy terms between citrate 1 and 2 are included because they are hydrogen-bonded to one another via an unusually short hydrogen bond between carboxylate / hydroxyl groups. No energy terms are included among HOH_591, and OgSer195, and O6' of the inhibitor. These atoms form a very short multi-centered hydrogen-bonding network.
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→7 Å
| ||||||||||||||||||||
| Refine LS restraints |
|
