 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1g72: CATALYTIC MECHANISM OF QUINOPROTEIN METHANOL DEHYDROGENASE: A THE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g72 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CATALYTIC MECHANISM OF QUINOPROTEIN METHANOL DEHYDROGENASE: A THEORETICAL AND X-RAY CRYSTALLOGRAPHIC INVESTIGATION | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | OXIDOREDUCTASE / Quinoprotein | |||||||||
| Function / homology |  Function and homology information Function and homology informationalcohol dehydrogenase (cytochrome c(L)) activity / methanol dehydrogenase (cytochrome c) / methanol oxidation / methanol metabolic process / alcohol dehydrogenase (NAD+) activity / outer membrane-bounded periplasmic space / calcium ion binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Methylophilus methylotrophus (bacteria) Methylophilus methylotrophus (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | |||||||||
 Authors Authors | Zheng, Y. / Xia, Z. / Chen, Z. / Bruice, T.C. / Mathews, F.S. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2001 Title: Catalytic mechanism of quinoprotein methanol dehydrogenase: A theoretical and x-ray crystallographic investigation. Authors: Zheng, Y.J. / Xia, Z.x. / Chen, Z.w. / Mathews, F.S. / Bruice, T.C. #1: Journal: J.Mol.Biol. / Year: 1996Title: DETERMINATION OF THE GENE SEQUENCE AND THE THREE-DIMENSIONAL STRUCTURE AT 2.4 ANGSTROMS RESOLUTION OF METHANOL DEHYDROGENASE FROM METHYLOPHILUS W3A1 Authors: Xia, Z. / Dai, W. / ZHANG, Y. / WHITE, S.A. / BOYD, G.D. / Mathews, F.S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g72.cif.gz | 264.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g72.ent.gz | 210 KB | Display | PDB format |
| PDBx/mmJSON format | 1g72.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g7/1g72ftp://data.pdbj.org/pub/pdb/validation_reports/g7/1g72 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4aahS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | METHANOL DEHYDROGENASE IS AN A2B2 TETRAMER. THE ASYMMETRIC UNIT CONTAINS THE TETRAMER, TWO PYRROLOQUINOLINE QUINONE COFACTORS (PQQ) AND 2 CALCIUM IONS. A NON-CRYSTALLOGRAPHIC TWO-FOLD AXIS RELATES THE TWO HALVES. |
-Components
| #1: Protein | Mass: 62702.828 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Methylophilus methylotrophus (bacteria) / Strain: W3A1 / References: UniProt: P38539, EC: 1.1.99.8#2: Protein | Mass: 7770.640 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Methylophilus methylotrophus (bacteria) / Strain: W3A1 / References: UniProt: P38540, EC: 1.1.99.8#3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Chemical | 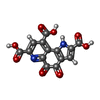  Mass: 330.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H6N2O8 Mass: 330.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H6N2O8#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 614 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 614 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.79 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: directly mixing / pH: 8.2 Details: PEG 8000, tris-HCl, Methanol, pH 8.2, Directly mixing, temperature 295.0K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 9.2 / Method: vapor diffusion / Details: Oubrie, A., (1999) J.Mol.Biol., 289, 319. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: BL-6B / Wavelength: 1 Å / Beamline: BL-6B / Wavelength: 1 Å |
| Detector | Type: WEISSENBERG / Detector: DIFFRACTOMETER / Date: Nov 1, 1995 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→40 Å / Num. all: 109884 / Num. obs: 108348 / % possible obs: 98.6 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 5 % / Biso Wilson estimate: 13.7 Å2 / Rmerge(I) obs: 0.091 / Net I/σ(I): 15.5 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.81 / Mean I/σ(I) obs: 1.9 / Num. unique all: 10453 / % possible all: 95.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4AAH Resolution: 1.9→100 Å / Isotropic thermal model: ISOTROPIC / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.5 Å2 | |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→100 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.97 Å
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.9 Å / Rfactor Rfree: 0.304 / Rfactor Rwork: 0.281 |
