 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4tpg: Selectivity mechanism of a bacterial homologue of the human drug ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4tpg | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Selectivity mechanism of a bacterial homologue of the human drug peptide transporters PepT1 and PepT2 | |||||||||||||||
 Components Components |
| |||||||||||||||
 Keywords Keywords | MEMBRANE PROTEIN / complex / secondary active transporter | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationdipeptide transmembrane transport / tripeptide transmembrane transport / tripeptide transmembrane transporter activity / peptide:proton symporter activity / dipeptide transmembrane transporter activity / identical protein binding / plasma membrane Similarity search - Function | |||||||||||||||
| Biological species |  Shewanella oneidensis MR-1 (bacteria) Shewanella oneidensis MR-1 (bacteria)synthetic construct (others) | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.91 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.91 Å | |||||||||||||||
 Authors Authors | Guettou, F. / Quistgaard, E.M. / Raba, M. / Moberg, P. / Low, C. / Nordlund, P. | |||||||||||||||
| Funding support |  Sweden, Sweden,  Singapore, 4items Singapore, 4items
| |||||||||||||||
 Citation Citation | Journal: Nat.Struct.Mol.Biol. / Year: 2014 Title: Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Authors: Guettou, F. / Quistgaard, E.M. / Raba, M. / Moberg, P. / Low, C. / Nordlund, P. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4tpg.cif.gz | 368 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4tpg.ent.gz | 306.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4tpg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tp/4tpgftp://data.pdbj.org/pub/pdb/validation_reports/tp/4tpg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4tphC  4tpjC  4lepS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 57224.152 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Shewanella oneidensis MR-1 (bacteria) / Gene: SO_1277 / Production host: #2: Protein/peptide | |   Type: Polypeptide / Class: Transport activator / Mass: 481.137 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: 5,6-DIHYDRO-BENZO[H]CINNOLIN-3-YLAMINE Type: Polypeptide / Class: Transport activator / Mass: 481.137 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) / References: 5,6-DIHYDRO-BENZO[H]CINNOLIN-3-YLAMINE#3: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Sugar | 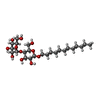  Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YM Type: D-saccharide / Mass: 510.615 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H46O11 / Comment: detergent*YMHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.05 Å3/Da / Density % sol: 69.61 % |
|---|---|
| Crystal grow | Temperature: 297 K / Method: vapor diffusion, sitting drop / pH: 4 / Details: 0.1 M phosphate citrate, 36% PEG300, 0.12 M ZnCl2 / PH range: 4 |
-Data collection
| Diffraction | Mean temperature: 80 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I02 / Wavelength: 0.9 Å / Beamline: I02 / Wavelength: 0.9 Å |
| Detector | Type: ADSC QUANTUM 1 / Detector: CCD / Date: Apr 20, 2013 |
| Radiation | Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 |
| Reflection | Resolution: 3.91→4.01 Å / Num. obs: 31871 / % possible obs: 90.6 % / Redundancy: 4.8 % / Net I/σ(I): 7.14 |
| Reflection shell | Resolution: 3.91→4.01 Å / Redundancy: 4.6 % / Mean I/σ(I) obs: 2.18 / % possible all: 39.3 |
- Processing
Processing
| Software | Name: PHENIX / Version: (phenix.refine: 1.8.2_1309) / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4LEP Resolution: 3.91→4.01 Å / SU ML: 0.55 / Cross valid method: NONE / σ(F): 1.37 / Phase error: 32.8 / Stereochemistry target values: ML Details: 52.30 percent of collected reflections collected used in refinement. The crystal suffered severe radiation damage and "died" during collection. Authors had to cut the data prior to ...Details: 52.30 percent of collected reflections collected used in refinement. The crystal suffered severe radiation damage and "died" during collection. Authors had to cut the data prior to refinement dues to bad quality. They also used the online anisotropy server (http://services.mbi.ucla.edu/anisoscale/) for data scaling.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.91→4.01 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
