 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fiw | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THREE-DIMENSIONAL STRUCTURE OF BETA-ACROSIN FROM RAM SPERMATOZOA | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE / anti-parallel beta-barrel | |||||||||
| Function / homology |  Function and homology information Function and homology informationacrosin / acrosome reaction / fucose binding / amidase activity / D-mannose binding / single fertilization / activation of adenylate cyclase activity / serine-type peptidase activity / acrosomal vesicle / serine-type endopeptidase activity / proteolysis Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.1 Å | |||||||||
 Authors Authors | Tranter, R. / Read, J.A. / Jones, R. / Brady, R.L. | |||||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 2000 Title: Effector sites in the three-dimensional structure of mammalian sperm beta-acrosin. Authors: Tranter, R. / Read, J.A. / Jones, R. / Brady, R.L. #1: Journal: To Be PublishedTitle: Three Dimensional Structure of Acrosin from Ram and Boar Spermatozoa Authors: Tranter, R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fiw.cif.gz | 76.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fiw.ent.gz | 55 KB | Display | PDB format |
| PDBx/mmJSON format | 1fiw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fi/1fiwftp://data.pdbj.org/pub/pdb/validation_reports/fi/1fiw | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | Heterodimer of chain A and L linked together by two disulphide bonds |
-Components
| #1: Protein | Mass: 32002.580 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 2440.680 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||
| #3: Polysaccharide | alpha-D-mannopyranose-(1-6)-beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1- ...alpha-D-mannopyranose-(1-6)-beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-[beta-L-fucopyranose-(1-6)]2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source | ||||
| #4: Chemical | 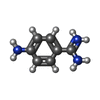  Mass: 136.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H10N3 Mass: 136.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H10N3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 191 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 191 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 56.15 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 4.6 Details: sodium acetate, sodium formate, p-aminobenzamidine, pH 4.6, VAPOR DIFFUSION, HANGING DROP, temperature 18K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 8.2 / Method: vapor diffusion | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.488 / Beamline: PX7.2 / Wavelength: 1.488 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 1, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. all: 23810 / Num. obs: 20601 / % possible obs: 93.7 % / Observed criterion σ(I): -3 / Redundancy: 4.8 % / Biso Wilson estimate: 40.2 Å2 / Rmerge(I) obs: 0.068 / Net I/σ(I): 15.3 |
| Reflection shell | Resolution: 2.1→2.2 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.298 / Num. unique all: 2710 / % possible all: 92.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→30 Å / SU B: 5.61319 / SU ML: 0.14893 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.20194 / ESU R Free: 0.18313 / Stereochemistry target values: Engh and Huber / Details: Maximum likelihood refinement
| ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.83 Å2
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→30 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 30 Å / σ(F): 0 / % reflection Rfree: 5.2 % | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
