登録情報 データベース : PDB / ID : 1etoタイトル THE CRYSTAL STRUCTURE OF E. COLI FIS MUTANT R71L FACTOR FOR INVERSION STIMULATION キーワード / / 機能・相同性 分子機能 ドメイン・相同性 構成要素
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / 生物種 Escherichia coli (大腸菌)手法 / 解像度 : 1.9 Å データ登録者 Cheng, Y.S. / Yang, W.Z. / Johnson, R.C. / Yuan, H.S. ジャーナル : J.Mol.Biol. / 年 : 2000タイトル : Structural analysis of the transcriptional activation on Fis: crystal structures of six Fis mutants with different activation properties.著者 : Cheng, Y.S. / Yang, W.Z. / Johnson, R.C. / Yuan, H.S. 履歴 登録 2000年4月13日 登録サイト / 処理サイト 改定 1.0 2000年10月11日 Provider / タイプ 改定 1.1 2008年4月27日 Group 改定 1.2 2011年7月13日 Group 改定 1.3 2021年11月3日 Group / カテゴリ / struct_ref_seq_difItem / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details改定 1.4 2024年2月7日 Group / カテゴリ / chem_comp_bond改定 1.5 2024年4月3日 Group / カテゴリ
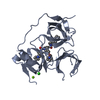
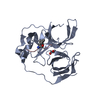
すべて表示 表示を減らす Remark 300 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 2 CHAINS THAT GIVE ONE ... THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 2 CHAINS THAT GIVE ONE BIOLOGICAL DIMER MOLECULE.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 X線回折 / 解像度: 1.9 Å
X線回折 / 解像度: 1.9 Å  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード
















 PDBj
PDBj 集合体
集合体
 分子量: 18.015 Da / 分子数: 115 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 115 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析