 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dr1: 2.2 ANGSTROMS CRYSTAL STRUCTURE OF CHICKEN LIVER DIHYDROFOLATE RE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dr1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | 2.2 ANGSTROMS CRYSTAL STRUCTURE OF CHICKEN LIVER DIHYDROFOLATE REDUCTASE COMPLEXED WITH NADP+ AND BIOPTERIN | ||||||
 Components Components | DIHYDROFOLATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtetrahydrofolate metabolic process / response to methotrexate / dihydrofolate metabolic process / dihydrofolate reductase / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / one-carbon metabolic process / NADP binding / mRNA binding / mitochondrion Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.2 Å X-RAY DIFFRACTION / Resolution: 2.2 Å | ||||||
 Authors Authors | Mctigue, M.A. / Davies /II, J.F. / Kaufman, B.T. / Xuong, N.-H. / Kraut, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1992 Title: Crystal structure of chicken liver dihydrofolate reductase complexed with NADP+ and biopterin. Authors: McTigue, M.A. / Davies 2nd., J.F. / Kaufman, B.T. / Kraut, J. #1: Journal: Biochemistry / Year: 1990Title: Crystal Structures of Recombinant Human Dihydrofolate Reductase Complexed with Folate and 5-Deazafolate Authors: Davies II, J.F. / Delcamp, T.J. / Prendergast, N.J. / Ashford, V.A. / Freisheim, J.H. / Kraut, J. #2: Journal: J.Biol.Chem. / Year: 1985Title: Refined Crystal Structure of Escherichia Coli and Chicken Liver Dihydrofolate Reductase Containing Bound Trimethoprim Authors: Matthews, D.A. / Bolin, J.T. / Burridge, J.M. / Filman, D.J. / Volz, K.W. / Kaufman, B.T. / Beddell, C.R. / Champness, J.N. / Stammers, D.K. / Kraut, J. #3: Journal: J.Biol.Chem. / Year: 1985Title: Dihydrofolate Reductase, the Stereochemistry of Inhibitor Selectivity Authors: Matthews, D.A. / Bolin, J.T. / Burridge, J.M. / Filman, D.J. / Volz, K.W. / Kraut, J. #4: Journal: J.Biol.Chem. / Year: 1982Title: Crystal Structure of Avian Dihydrofolate Reductase Containing Phenyltriazine and Nadph Authors: Volz, K.W. / Matthews, D.A. / Alden, R.A. / Freer, S.T. / Hansch, C. / Kaufman, B.T. / Kraut, J. #5: Journal: Biochemistry / Year: 1980Title: Primary Structure of Chicken Liver Dihydrofolate Reductase Authors: Kumar, A.A. / Blankenship, D.T. / Kaufman, B.T. / Freisheim, J.H. | ||||||
| History |
| ||||||
| Remark 650 | HELIX RESIDUES 21-26 (NLPWPP) FORM LEFT-HANDED POLYPROLINE HELIX. RESIDUES 108 AND 109 PARTICIPATE ...HELIX RESIDUES 21-26 (NLPWPP) FORM LEFT-HANDED POLYPROLINE HELIX. RESIDUES 108 AND 109 PARTICIPATE IN BOTH HELIX EP AND BETA STRAND E. | ||||||
| Remark 700 | SHEET RESIDUES 110 AND 111 FORM A BETA-BULGE IN STRAND E. RESIDUES 115 AND 116 FORM A BETA-BULGE IN ...SHEET RESIDUES 110 AND 111 FORM A BETA-BULGE IN STRAND E. RESIDUES 115 AND 116 FORM A BETA-BULGE IN STRAND E. RESIDUES 172 AND 175 PARTICIPATE IN BOTH TIGHT-TURN 8 AND BETA STRANDS 7 AND 9. TIGHT TURN 7 (RESIDUES 162-165) DISRUPT LAST STRAND OF SHEET INTO 2 STRANDS 8 AND 9. THIS STRAND IS CONTINUOUS IN E. COLI. A BETA BULGE IS PRESENT HERE IN L. CASEI. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dr1.cif.gz | 57.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dr1.ent.gz | 40.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1dr1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dr/1dr1ftp://data.pdbj.org/pub/pdb/validation_reports/dr/1dr1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES 21-26 (NLPWPP) FORM LEFT-HANDED POLYPROLINE HELIX. 2: RESIDUE TYR 31 EXISTS IN TWO CONFORMATIONS. / 3: CIS PROLINE - PRO 66 4: RESIDUES 108 AND 109 PARTICIPATE IN BOTH HELIX EP AND BETA STRAND E. 5: RESIDUES 110 AND 111 FORM A BETA-BULGE IN STRAND E. / 6: RESIDUES 115 AND 116 FORM A BETA-BULGE IN STRAND E. 7: PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION GLY 116 - GLY 117 9.948 8: TIGHT TURN 7 (RESIDUES 162-165) DISRUPT LAST STRAND OF SHEET INTO 2 STRANDS 8 AND 9. THIS STRAND IS CONTINUOUS IN E. COLI. A BETA BULGE IS PRESENT HERE IN L. CASEI. 9: RESIDUES 172 AND 175 PARTICIPATE IN BOTH TIGHT-TURN 8 AND BETA STRANDS 7 AND 9. 10: RESIDUE 200 (CALCIUM ION) LIES ON THE CRYSTALLOGRAPHIC TWO-FOLD AT ONE-HALF OCCUPANCY. | ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 21679.932 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #3: Chemical | ChemComp-NAP /   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 |
| #4: Chemical | ChemComp-HBI / 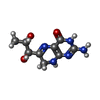  Mass: 239.231 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H13N5O3 Mass: 239.231 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H13N5O3 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.62 Å3/Da / Density % sol: 53.02 % | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.5 / Method: vapor diffusion | |||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.2 Å / Num. obs: 10688 / % possible obs: 91.2 % / Rmerge(I) obs: 0.022 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor obs: 0.14 / Highest resolution: 2.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2.2 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Rfactor obs: 0.14 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 22.7 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: p_angle_d / Dev ideal: 0.04 |
