 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dct | ||||||
|---|---|---|---|---|---|---|---|
| Title | DNA (CYTOSINE-5) METHYLASE FROM HAEIII COVALENTLY BOUND TO DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/DNA / ENZYME / CYTOSINE METHYLASE / TRANSFERASE-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA (cytosine-5-)-methyltransferase / DNA (cytosine-5-)-methyltransferase activity / DNA restriction-modification system / negative regulation of gene expression via chromosomal CpG island methylation / methylation / DNA binding / ATP binding Similarity search - Function | ||||||
| Biological species |  Haemophilus influenzae biotype aegyptius (bacteria) Haemophilus influenzae biotype aegyptius (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Reinisch, K.M. / Chen, L. / Verdine, G.L. / Lipscomb, W.N. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1995 Title: The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Authors: Reinisch, K.M. / Chen, L. / Verdine, G.L. / Lipscomb, W.N. #1: Journal: J.Mol.Biol. / Year: 1994Title: Crystallization and Prelimanary Crystallographic Analysis of a DNA (Cytosine-5) -Methyltransferase from Haemophilus Aegyptius Bound Covalently to DNA Authors: Reinisch, K.M. / Chen, L. / Verdine, G.L. / Lipscomb, W.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dct.cif.gz | 185.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dct.ent.gz | 143 KB | Display | PDB format |
| PDBx/mmJSON format | 1dct.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dc/1dctftp://data.pdbj.org/pub/pdb/validation_reports/dc/1dct | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 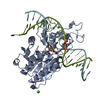
| ||||||||||
| 2 | 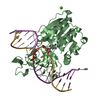
| ||||||||||
| Unit cell |
| ||||||||||
| Details | THIS FILE CONTAINS 2 PROTEIN-DNA COMPLEXES IN THE ASYMMETRIC UNIT. PROTEIN MONOMER A IS COVALENTLY BOUND TO A DNA DUPLEX CONSISTING OF CHAINS F AND M, AND PROTEIN MONOMER B IS COVALENTLY LINKED TO A DUPLEX CONSISTING OF CHAINS G AND N. THERE ARE TWO SPECIAL NUCLEOTIDE BASES INCORPORATED INTO EACH DNA DUPLEX - ONE IRREGULAR BASE IN EACH CHAIN. ONE OF THESE BASES IS A CYTOSINE METHYLATED AT THE 5-POSITION, AND THE OTHER (DESCRIBED IN MORE DETAIL BELOW) IS USED TO COVALENTLY LINK THE DNA TO THE PROTEIN. THE DNA AT THE RECOGNITION SITE IS VERY DISTORTED: THERE IS AN EXTRAHELICAL CYTOSINE (THE MODIFIED ONE COVALENTLY LINKED TO THE PROTEIN VIA CYS 71: +C F 10 AND +C G 10) AND BASE PAIRING IN THE DNA RECOGNITION SEQUENCE IS REORGANIZED. THERE ARE ALSO TWO CA+2 IONS IN THE ASYMMETRIC UNIT. |
-Components
| #1: DNA chain | Mass: 5559.653 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: DNA chain | Mass: 5553.590 Da / Num. of mol.: 2 / Source method: obtained synthetically #3: Protein | Mass: 37093.559 Da / Num. of mol.: 2 / Source method: isolated from a natural source Source: (natural) Haemophilus influenzae biotype aegyptius (bacteria)Species: Haemophilus influenzae / Strain: biotype aegyptius / References: UniProt: P20589, EC: 2.1.1.73 #4: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CaHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: pH 6.50, VAPOR DIFFUSION, HANGING DROP, temperature 277.00K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 113 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.7 Å / Num. all: 129524 / Num. obs: 26866 / % possible obs: 95 % / Rmerge(I) obs: 0.076 |
| Reflection | *PLUS Highest resolution: 2.7 Å / % possible obs: 95 % / Observed criterion σ(I): 1 / Num. measured all: 129524 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→10 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 10 Å / σ(F): 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
