 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dck | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF UNPHOSPHORYLATED FIXJ-N COMPLEXED WITH MN2+ | ||||||
 Components Components | TRANSCRIPTIONAL REGULATORY PROTEIN FIXJ | ||||||
 Keywords Keywords | TRANSCRIPTION / DOUBLY WOUND FIVE-STRANDED BETA/ALPHA FOLD / NITROGEN FIXATION REGULATION | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA-binding transcription activator activity / phosphorelay signal transduction system / protein-DNA complex / transcription cis-regulatory region binding / positive regulation of DNA-templated transcription / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Sinorhizobium meliloti (bacteria) Sinorhizobium meliloti (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2 Å | ||||||
 Authors Authors | Gouet, P. / Fabry, B. / Guillet, V. / Birck, C. / Mourey, L. / Kahn, D. / Samama, J.P. | ||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 1999 Title: Structural transitions in the FixJ receiver domain. Authors: Gouet, P. / Fabry, B. / Guillet, V. / Birck, C. / Mourey, L. / Kahn, D. / Samama, J.P. #1: Journal: To be PublishedTitle: Conformational changes induced by phosphorylation of the FixJ receiver domain Authors: Birck, C. / Mourey, L. / Gouet, P. / Fabry, B. / Schumacher, J. / Rousseau, P. / Kahn, D. / Samama, J.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dck.cif.gz | 65.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dck.ent.gz | 45.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1dck.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dc/1dckftp://data.pdbj.org/pub/pdb/validation_reports/dc/1dck | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
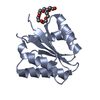
| 1 | 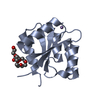
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13880.008 Da / Num. of mol.: 2 / Fragment: N-TERMINAL DOMAIN (RESIDUES 1-126) / Mutation: T2Q, A125L Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sinorhizobium meliloti (bacteria) / Plasmid: PT7-7 / Production host: #2: Chemical |   Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mn#3: Chemical | ChemComp-15P / | 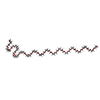  Mass: 1529.829 Da / Num. of mol.: 1 / Fragment: POLYETHYLENE GLYCOL, 19 ATOMS / Source method: obtained synthetically / Formula: C69H140O35 / Comment: precipitant*YM Mass: 1529.829 Da / Num. of mol.: 1 / Fragment: POLYETHYLENE GLYCOL, 19 ATOMS / Source method: obtained synthetically / Formula: C69H140O35 / Comment: precipitant*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 94 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 35 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: PEG1500, pH 7, VAPOR DIFFUSION, HANGING DROP, temperature 4K | |||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 30 % | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.95 / Beamline: ID14-4 / Wavelength: 0.95 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jan 1, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 |
| Reflection | Resolution: 2→30 Å / Num. all: 24325 / Num. obs: 13062 / % possible obs: 87 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 1.9 % / Biso Wilson estimate: 17.1 Å2 / Rmerge(I) obs: 0.051 / Net I/σ(I): 15.6 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.196 / % possible all: 57.7 |
| Reflection | *PLUS Highest resolution: 2 Å / Lowest resolution: 30 Å / % possible obs: 87 % / Num. measured all: 24325 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2→14.45 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 701624.7 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 26.82 Å2 / ksol: 0.349 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→14.45 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.12 Å / Rfactor Rfree error: 0.028 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 7 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 25.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.308 / % reflection Rfree: 7.5 % / Rfactor Rwork: 0.25 |
