 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dc1: RESTRICTION ENZYME BSOBI/DNA COMPLEX STRUCTURE: ENCIRCLEMENT OF T... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dc1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | RESTRICTION ENZYME BSOBI/DNA COMPLEX STRUCTURE: ENCIRCLEMENT OF THE DNA AND HISTIDINE-CATALYZED HYDROLYSIS WITHIN A CANONICAL RESTRICTION ENZYME FOLD | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE/DNA / PROTEIN-DNA COMPLEX / RESTRICTION ENDONUCLEASE / THERMOPHILIC ENZYME / DEGENERATE DNA RECOGNITION / HYDROLASE-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationtype II site-specific deoxyribonuclease / type II site-specific deoxyribonuclease activity / DNA restriction-modification system / DNA binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MIR / Resolution: 1.7 Å X-RAY DIFFRACTION / MIR / Resolution: 1.7 Å | ||||||
 Authors Authors | van der Woerd, M.J. / Pelletier, J.J. / Xu, S.-Y. / Friedman, A.M. | ||||||
 Citation Citation | Journal: Structure / Year: 2001 Title: Restriction enzyme BsoBI-DNA complex: a tunnel for recognition of degenerate DNA sequences and potential histidine catalysis. Authors: van der Woerd, M.J. / Pelletier, J.J. / Xu, S. / Friedman, A.M. #1: Journal: Mol.Gen.Genet. / Year: 1996Title: Cloning and sequence comparison of AvaI and BsoBI restriction modification systems Authors: Ruan, H. / Lunnen, K.D. / Scott, M.E. / Moran, L.S. / Slatko, B.E. / Pelletier, J.J. / Hess, E.J. / Benner II, J. / Wilson, G.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dc1.cif.gz | 161.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dc1.ent.gz | 124.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1dc1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dc/1dc1ftp://data.pdbj.org/pub/pdb/validation_reports/dc/1dc1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3965.609 Da / Num. of mol.: 2 / Source method: obtained synthetically Details: DNA SEQUENCE DESIGNED FOR CRYSTALLIZATION BASED ON THE CENTRAL RECOGNITION SEQUENCE OF BACILLUS STEAROTHERMOPHILUS #2: Protein | Mass: 36742.793 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus stearothermophilus (bacteria)Production host: References: UniProt: P70985, type II site-specific deoxyribonuclease #3: Chemical | ChemComp-DIO / 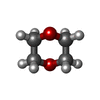  Mass: 88.105 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C4H8O2 Mass: 88.105 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C4H8O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 474 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 474 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41.2 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.6 Details: EQUILIBRATION AGAINST RESERVOIR OF 20% (V/V) DIOXANE, 2% (V/V) ETHYLENE GLYCOL AND 5 MM DTT. DROPS WERE FORMED BY MIXING 2.5 UL PROTEIN-DNA COMPLEX (10 MG/ML PROTEIN; 1.1 FOLD MOLAR EXCESS ...Details: EQUILIBRATION AGAINST RESERVOIR OF 20% (V/V) DIOXANE, 2% (V/V) ETHYLENE GLYCOL AND 5 MM DTT. DROPS WERE FORMED BY MIXING 2.5 UL PROTEIN-DNA COMPLEX (10 MG/ML PROTEIN; 1.1 FOLD MOLAR EXCESS DNA) IN BUFFER (20 MM TRIS-HCL PH 7.6, 300 MM NACL, 0.1 MM EDTA, 1 MM DTT, 0.02% NA AZIDE) WITH 2.5 UL DISTILLED WATER, 2.5 UL RESERVOIR SOLUTION AND 1.5 UL 350 MM N-HEPTYL-B-D-GLUCOSIDE, VAPOR DIFFUSION, HANGING DROP, temperature 293.0K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Sep 1, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→30 Å / Num. all: 77509 / Num. obs: 77509 / % possible obs: 99.4 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 3.7 % / Biso Wilson estimate: 20.9 Å2 / Rmerge(I) obs: 0.059 / Net I/σ(I): 24.5 |
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 2.2 % / Rmerge(I) obs: 0.519 / Mean I/σ(I) obs: 2.1 / % possible all: 95.4 |
| Reflection | *PLUS |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 1.7→8 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Cross valid method: THROUGHOUT / σ(F): 2 Stereochemistry target values: ENGH & HUBER AND PARKINSON ET AL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.33 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.197 Å / Luzzati d res low obs: 8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Rms dev position: 0.0991 Å / Weight Biso : 300 / Weight position: 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.78 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 8 Å / σ(F): 2 / % reflection Rfree: 5.1 % / Rfactor obs: 0.19 / Rfactor Rwork: 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_deg / Dev ideal: 1.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.45 / % reflection Rfree: 5 % / Rfactor Rwork: 0.402 |
