 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1dam: DETHIOBIOTIN SYNTHETASE COMPLEXED WITH DETHIOBIOTIN, ADP, INORGAN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dam | ||||||
|---|---|---|---|---|---|---|---|
| Title | DETHIOBIOTIN SYNTHETASE COMPLEXED WITH DETHIOBIOTIN, ADP, INORGANIC PHOSPHATE AND MAGNESIUM | ||||||
 Components Components | PROTEIN (DETHIOBIOTIN SYNTHETASE) | ||||||
 Keywords Keywords | LIGASE / BIOTIN BIOSYNTHESIS / MAGNESIUM / ATP-BINDING / PHOSPHORYL TRANSFER | ||||||
| Function / homology |  Function and homology information Function and homology informationdethiobiotin synthase / dethiobiotin synthase activity / biotin biosynthetic process / magnesium ion binding / protein homodimerization activity / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 1.8 Å X-RAY DIFFRACTION / OTHER / Resolution: 1.8 Å | ||||||
 Authors Authors | Kaeck, H. / Sandmark, J. / Gibson, K.J. / Schneider, G. / Lindqvist, Y. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1998 Title: Crystal structure of two quaternary complexes of dethiobiotin synthetase, enzyme-MgADP-AlF3-diaminopelargonic acid and enzyme-MgADP-dethiobiotin-phosphate; implications for catalysis. Authors: Kack, H. / Sandmark, J. / Gibson, K.J. / Schneider, G. / Lindqvist, Y. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1998Title: Snapshot of a Phosphorylated Substrate Intermediate by Kinetic Crystallography Authors: Kaeck, H. / Gibson, K.J. / Schneider, G. / Lindqvist, Y. #2: Journal: Structure / Year: 1994Title: Crystal Structure of an ATP-Dependent Carboxylase, Dethiobiotin Synthetase, at 1.65 A Resolution Authors: Huang, W. / Lindqvist, Y. / Schneider, G. / Gibson, K.J. / Flint, D. / Lorimer, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dam.cif.gz | 62.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dam.ent.gz | 44.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1dam.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1dam_validation.pdf.gz | 806.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1dam_full_validation.pdf.gz | 808.3 KB | Display | |
| Data in XML | 1dam_validation.xml.gz | 13 KB | Display | |
| Data in CIF | 1dam_validation.cif.gz | 18.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/da/1damftp://data.pdbj.org/pub/pdb/validation_reports/da/1dam | HTTPS FTP |
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 24028.289 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|
-Non-polymers , 5 types, 204 molecules 


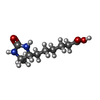

| #2: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-PO4 / |  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#4: Chemical | ChemComp-ADP / |  Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#5: Chemical | ChemComp-DTB / |  Mass: 214.262 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H18N2O3 Mass: 214.262 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H18N2O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 199 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.16 Å3/Da / Density % sol: 34 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 | ||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Jul 1, 1996 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→30 Å / Num. obs: 17046 / % possible obs: 90.6 % / Redundancy: 2.5 % / Rsym value: 0.044 / Net I/σ(I): 18.1 |
| Reflection shell | Resolution: 1.8→1.86 Å / Mean I/σ(I) obs: 3.3 / Rsym value: 0.337 / % possible all: 86.1 |
| Reflection | *PLUS Num. measured all: 42816 / Rmerge(I) obs: 0.044 |
| Reflection shell | *PLUS Highest resolution: 1.8 Å / % possible obs: 86.1 % / Rmerge(I) obs: 0.337 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.8→20 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / σ(F): 0 / % reflection Rfree: 4.7 % / Rfactor obs: 0.18 / Rfactor Rwork: 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 15.6 Å2 |
