 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1d37: INFLUENCE OF AGLYCONE MODIFICATIONS ON THE BINDING OF ANTHRACYCLI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1d37 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | INFLUENCE OF AGLYCONE MODIFICATIONS ON THE BINDING OF ANTHRACYCLINE DRUGS TO DNA: THE MOLECULAR STRUCTURE OF IDARUBICIN AND 4-O-DEMETHYL-11-DEOXYDOXORUBICIN COMPLEXED TO D(CGATCG) | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / RIGHT HANDED DNA / DOUBLE HELIX / COMPLEXED WITH DRUG | Function / homology | 1-O-DEMETHYL-6-DEOXYDOXORUBICIN / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / Resolution: 1.8 Å X-RAY DIFFRACTION / Resolution: 1.8 Å  Authors AuthorsGao, Y.-G. / Wang, A.H.-J. |  CitationJournal: Anti-Cancer Drug Des. / Year: 1991 CitationJournal: Anti-Cancer Drug Des. / Year: 1991Title: Influence of aglycone modifications on the binding of anthracycline drugs to DNA: the molecular structure of idarubicin and 4-O-demethyl-11-deoxydoxorubicin complexed to d(CGATCG). Authors: Gao, Y.G. / Wang, A.H. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1991Title: Facile Formation of Crosslinked Adduct between DNA and the Daunorubicin Derivative MAR70 Mediated by Formaldehyde: Molecular Structure of the MAR70-d(CGTnACG) Covalent Adduct Authors: Gao, Y.-G. / Liaw, Y.-C. / Li, Y.-K. / Van Der Marel, G.A. / Van Boom, J.H. / Wang, A.H.-J. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1d37.cif.gz | 15.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1d37.ent.gz | 8.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1d37.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d3/1d37ftp://data.pdbj.org/pub/pdb/validation_reports/d3/1d37 | HTTPS FTP |
|---|
-Related structure data





-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 1809.217 Da / Num. of mol.: 1 / Source method: obtained synthetically |
|---|---|
| #2: Chemical | ChemComp-DM4 / 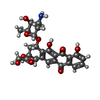  Mass: 513.493 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C26H27NO10 Mass: 513.493 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C26H27NO10 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 42 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 42 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.92 Å3/Da / Density % sol: 57.81 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 6 / Details: pH 6.00, VAPOR DIFFUSION, temperature 298.00K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion / pH: 6 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU AFC-5R / Detector: DIFFRACTOMETER |
| Radiation | Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 1.8 Å / Num. obs: 1247 / Observed criterion σ(F): 2 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Observed criterion σ(F): 2 |
- Processing
Processing
| Software | Name: NUCLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Highest resolution: 1.8 Å / σ(F): 2 /
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine Biso |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 1.8 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / σ(F): 2 / Rfactor obs: 0.179 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: n_bond_d / Dev ideal: 0.025 |
