 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-258d: FACTORS AFFECTING SEQUENCE SELECTIVITY ON NOGALAMYCIN INTERCALATI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 258d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | FACTORS AFFECTING SEQUENCE SELECTIVITY ON NOGALAMYCIN INTERCALATION: THE CRYSTAL STRUCTURE OF D(TGTACA)-NOGALAMYCIN | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / B-DNA / DOUBLE HELIX / COMPLEXED WITH DRUG | Function / homology | ACETATE ION / NOGALAMYCIN / SPERMINE / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.58 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.58 Å  Authors AuthorsSmith, C.K. / Brannigan, J.A. / Moore, M.H. |  CitationJournal: J.Mol.Biol. / Year: 1996 CitationJournal: J.Mol.Biol. / Year: 1996Title: Factors affecting DNA sequence selectivity of nogalamycin intercalation: the crystal structure of d(TGTACA)2-nogalamycin2. Authors: Smith, C.K. / Brannigan, J.A. / Moore, M.H. #1: Journal: Biochemistry / Year: 1995Title: DNA-Nogalamycin Interactions: The Crystal Structure of d(TGATCA) Complexed with Nogalamycin Authors: Smith, C.K. / Davies, G.J. / Dodson, E.J. / Moore, M.H. #2: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: DNA-Drug Refinement: A Comparison of the Programs NUCLSQ, PROLSQ, SHELXL93 and X-PLOR, Using the Low Temperature d(TGATCA)-Nogalamycin Structure Authors: Schuerman, G.S. / Smith, C.K. / Turkenburg, J.P. / Dettmar, A.N. / Van Meervelt, L. / Moore, M.H. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 258d.cif.gz | 32.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb258d.ent.gz | 23.4 KB | Display | PDB format |
| PDBx/mmJSON format | 258d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/58/258dftp://data.pdbj.org/pub/pdb/validation_reports/58/258d | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  182dS S: Starting model for refinement |
|---|---|
| Similar structure data |


-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 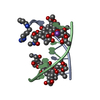
| ||||||||
| 2 | 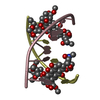
| ||||||||
| Unit cell |
|
-Components
-DNA chain , 1 types, 4 molecules ABCD
| #1: DNA chain | Mass: 1808.229 Da / Num. of mol.: 4 / Source method: obtained synthetically |
|---|
-Non-polymers , 5 types, 105 molecules 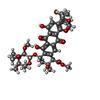
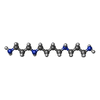



| #2: Chemical | ChemComp-NGM /  Mass: 787.803 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C39H49NO16 / Comment: antitumor, antibiotic*YM Mass: 787.803 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C39H49NO16 / Comment: antitumor, antibiotic*YM#3: Chemical | ChemComp-SPM / |  Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4 Mass: 202.340 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H26N4#4: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#5: Chemical |  Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.17 Å3/Da / Density % sol: 61.19 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: pH 6.50, VAPOR DIFFUSION, SITTING DROP, temperature 277.00K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X31 / Beamline: X31 |
| Detector | Type: HENDRIX-LENTFER / Detector: IMAGE PLATE / Date: Jul 1, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 1.58→10.69 Å / Num. obs: 13148 / % possible obs: 94 % / Observed criterion σ(I): 1 / Redundancy: 6.3 % / Rmerge(I) obs: 0.046 |
| Reflection shell | Resolution: 1.58→1.66 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.196 / % possible all: 69.5 |
| Reflection | *PLUS Highest resolution: 1.58 Å / Lowest resolution: 10.69 Å |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: NDB ENTRY DDF049 (PDB: 182D) Resolution: 1.58→10.69 Å / Num. parameters: 3310 / Num. restraintsaints: 12049 / Cross valid method: FREE R-VALUE / σ(F): 0 / Stereochemistry target values: TAYLOR AND KENNARD / Details: ESTIMATED COORDINATE ERROR (A) : 0.12A
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET'S PRINCIPLE | |||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.78 Å2 | |||||||||||||||||||||||||||||||||
| Refine Biso |
| |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.58→10.69 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-93 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.58 Å / Lowest resolution: 10.69 Å / σ(F): 0 / % reflection Rfree: 10 % | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_plane_restr / Dev ideal: 0.089 |
