 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-482d: RELEASE OF THE CYANO MOIETY IN THE CRYSTAL STRUCTURE OF N-CYANOME... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 482d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | RELEASE OF THE CYANO MOIETY IN THE CRYSTAL STRUCTURE OF N-CYANOMETHYL-N-(2-METHOXYETHYL)-DAUNOMYCIN COMPLEXED WITH D(CGATCG) | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / RIGHT HANDED DNA / DOUBLE HELIX / COMPLEXED WITH DRUG / DEOXYRIBONUCLEIC ACID | Function / homology | N-HYDROXYMETHYL-N-(2-METHOXYETHYL)-DAUNOMYCIN / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.54 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.54 Å  Authors AuthorsSaminadin, P. / Dautant, A. / Mondon, M. / Langlois D'Estaintot, B. / Courseille, C. / Precigoux, G. |  CitationJournal: Eur.J.Biochem. / Year: 2000 CitationJournal: Eur.J.Biochem. / Year: 2000Title: Release of the cyano moiety in the crystal structure of N-cyanomethyl-N-(2-methoxyethyl)-daunomycin complexed with d(CGATCG). Authors: Saminadin, P. / Dautant, A. / Mondon, M. / Langlois D'estaintot, B. / Courseille, C. / Precigoux, G. #1: Journal: Eur.J.Biochem. / Year: 1998Title: Degradation of the Morpholino Ring in the Crystal Structure of Cyanomorpholinodoxorubicin Authors: Ettorre, A. / Cirilli, M. / Ughetto, G. #2: Journal: Biochemistry / Year: 1990Title: Structural Comparison of Anticancer Drug-DNA Complexes. Adriamycin and Daunomycin Authors: Frederick, C.A. / Williams, L.D. / Ughetto, G. / Van Der Marel, G.A. / Van Boom, J.H. / Rich, A. / Wang, A.H.-J. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 482d.cif.gz | 15 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb482d.ent.gz | 8.4 KB | Display | PDB format |
| PDBx/mmJSON format | 482d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/82/482dftp://data.pdbj.org/pub/pdb/validation_reports/82/482d | HTTPS FTP |
|---|
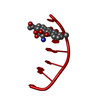
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 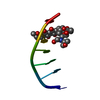
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 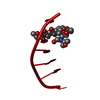
| ||||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 1809.217 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: COMPLEXED WITH (2-METHOXYETHYL)-DAUNOMYCIN |
|---|---|
| #2: Chemical | ChemComp-DM9 / 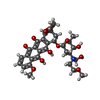  Mass: 617.641 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H39NO12 Mass: 617.641 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H39NO12 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 25 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 25 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | N-CYANOMETHYL-N-(2-METHOXYETHYL)-DAUNOMYCIN WAS USED IN CRYSTALLIZATION SET-UP. DEGRADATION OF THIS ...N-CYANOMETHY |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 44.5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: CRYSTALS WERE OBTAINED FROM A SOLUTION THAT CONTAINED MPD, LICL, MGCL2, SODIUM CACODYLATE, COBALT HEXAMINE, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR571 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 15, 1999 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.54→12.6 Å / Num. obs: 3327 / % possible obs: 95.5 % / Observed criterion σ(I): 0 / Redundancy: 3.9 % / Biso Wilson estimate: 15.3 Å2 / Rmerge(I) obs: 0.046 / Rsym value: 0.046 / Net I/σ(I): 9.6 |
| Reflection shell | Resolution: 1.54→1.57 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.158 / Mean I/σ(I) obs: 3.9 / Rsym value: 0.158 / % possible all: 92.3 |
| Reflection shell | *PLUS % possible obs: 92.3 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: SEE REMARK 1 REFERENCE 2 Resolution: 1.54→12.6 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.54→12.6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.54→1.58 Å / Total num. of bins used: 15
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
