 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cxz: CRYSTAL STRUCTURE OF HUMAN RHOA COMPLEXED WITH THE EFFECTOR DOMAI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cxz | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN RHOA COMPLEXED WITH THE EFFECTOR DOMAIN OF THE PROTEIN KINASE PKN/PRK1 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | SIGNALING PROTEIN / PROTEIN-PROTEIN COMPLEX / ANTIPARALLEL COILED-COIL | ||||||
| Function / homology |  Function and homology information Function and homology informationepithelial cell migration / protein kinase C / diacylglycerol-dependent serine/threonine kinase activity / alpha-beta T cell lineage commitment / aortic valve formation / positive regulation of lipase activity / endothelial tube lumen extension / renal system process / skeletal muscle satellite cell migration / positive regulation of vascular associated smooth muscle contraction ...epithelial cell migration / protein kinase C / diacylglycerol-dependent serine/threonine kinase activity / alpha-beta T cell lineage commitment / aortic valve formation / positive regulation of lipase activity / endothelial tube lumen extension / renal system process / skeletal muscle satellite cell migration / positive regulation of vascular associated smooth muscle contraction / angiotensin-mediated vasoconstriction involved in regulation of systemic arterial blood pressure / SLIT2:ROBO1 increases RHOA activity / RHO GTPases Activate Rhotekin and Rhophilins / Roundabout signaling pathway / bone trabecula morphogenesis / negative regulation of intracellular steroid hormone receptor signaling pathway / Axonal growth inhibition (RHOA activation) / Axonal growth stimulation / cleavage furrow formation / regulation of neural precursor cell proliferation / regulation of osteoblast proliferation / regulation of modification of postsynaptic actin cytoskeleton / forebrain radial glial cell differentiation / regulation of cell motility / mitotic cleavage furrow formation / apical junction assembly / negative regulation of cell migration involved in sprouting angiogenesis / B cell apoptotic process / beta selection / cell junction assembly / histone H3T11 kinase activity / establishment of epithelial cell apical/basal polarity / cellular response to chemokine / negative regulation of motor neuron apoptotic process / regulation of systemic arterial blood pressure by endothelin / negative regulation of oxidative phosphorylation / regulation of germinal center formation / regulation of immunoglobulin production / regulation of modification of postsynaptic structure / RHO GTPases Activate ROCKs / RHO GTPases activate CIT / negative regulation of cell size / regulation of androgen receptor signaling pathway / Sema4D induced cell migration and growth-cone collapse / PCP/CE pathway / RHO GTPases activate KTN1 / positive regulation of alpha-beta T cell differentiation / positive regulation of podosome assembly / apolipoprotein A-I-mediated signaling pathway / wound healing, spreading of cells / Sema4D mediated inhibition of cell attachment and migration / hyperosmotic response / motor neuron apoptotic process / positive regulation of leukocyte adhesion to vascular endothelial cell / odontogenesis / PI3K/AKT activation / Wnt signaling pathway, planar cell polarity pathway / ossification involved in bone maturation / regulation of focal adhesion assembly / androgen receptor signaling pathway / negative chemotaxis / nuclear androgen receptor binding / RHOB GTPase cycle / EPHA-mediated growth cone collapse / apical junction complex / negative regulation of B cell proliferation / stress fiber assembly / myosin binding / positive regulation of cytokinesis / regulation of neuron projection development / RHOC GTPase cycle / cellular response to cytokine stimulus / cerebral cortex cell migration / B cell homeostasis / ERBB2 Regulates Cell Motility / positive regulation of protein serine/threonine kinase activity / cleavage furrow / semaphorin-plexin signaling pathway / negative regulation of cell-substrate adhesion / ficolin-1-rich granule membrane / RHOA GTPase cycle / mitotic spindle assembly / positive regulation of T cell migration / endothelial cell migration / skeletal muscle tissue development / Rho protein signal transduction / spleen development / RHO GTPases activate PKNs / GPVI-mediated activation cascade / PTK6 Regulates RHO GTPases, RAS GTPase and MAP kinases / negative regulation of reactive oxygen species biosynthetic process / positive regulation of stress fiber assembly / cytoplasmic microtubule organization / RAC1 GTPase cycle / EPHB-mediated forward signaling / positive regulation of neuron differentiation / substrate adhesion-dependent cell spreading / substantia nigra development / post-translational protein modification / regulation of cell migration Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.2 Å X-RAY DIFFRACTION / Resolution: 2.2 Å | ||||||
 Authors Authors | Maesaki, R. / Ihara, K. / Shimizu, T. / Kuroda, S. / Kaibuchi, K. / Hakoshima, T. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 1999 Title: The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Authors: Maesaki, R. / Ihara, K. / Shimizu, T. / Kuroda, S. / Kaibuchi, K. / Hakoshima, T. #1: Journal: J.Struct.Biol. / Year: 1999Title: Biochemical and crystallographic characterization of a Rho effector domain of the protein serine/threonine kinase N in a complex with RhoA Authors: Maesaki, R. / Shimizu, T. / Ihara, K. / Kuroda, S. / Kaibuchi, K. / Hakoshima, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cxz.cif.gz | 69.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cxz.ent.gz | 50.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1cxz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cx/1cxzftp://data.pdbj.org/pub/pdb/validation_reports/cx/1cxz | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj


















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 20596.615 Da / Num. of mol.: 1 / Fragment: RESIDUES 1 - 181 / Mutation: G14V Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PRSET B / Production host:  |
|---|---|
| #2: Protein | Mass: 9811.257 Da / Num. of mol.: 1 / Fragment: RESIDUES 13 - 98 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PGET-2T / Production host: |
| #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #4: Chemical | ChemComp-GSP / 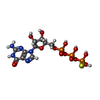  Mass: 539.246 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3S Mass: 539.246 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3S |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.75 Å3/Da / Density % sol: 44 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: microbatch / pH: 6.5 / Details: PEG 300, pH 6.5, MICROBATCH, temperature 277K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: batch method / Details: Maesaki, R., (1999) J.Struct.Biol., 126, 166. | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 90 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-C / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Jan 5, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→49.8 Å / Num. all: 696854 / Num. obs: 17568 / % possible obs: 96 % / Observed criterion σ(I): 1 / Biso Wilson estimate: 32.8 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 24 |
| Reflection shell | Resolution: 2.2→2.28 Å / Rmerge(I) obs: 0.312 / % possible all: 86 |
| Reflection | *PLUS % possible obs: 96 % / Num. measured all: 696854 / Rmerge(I) obs: 0.09 |
| Reflection shell | *PLUS Highest resolution: 2.2 Å / % possible obs: 86 % / Mean I/σ(I) obs: 2.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.2→49.8 Å / σ(F): 2 / Stereochemistry target values: PROTEIN.PARAM
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→49.8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 49.8 Å / σ(F): 2 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: c_dihedral_angle_deg / Dev ideal: 22.45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 2.28 Å / Rfactor Rfree: 0.359 / Rfactor obs: 0.315 |
