 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cje | ||||||
|---|---|---|---|---|---|---|---|
| Title | ADRENODOXIN FROM BOVINE | ||||||
 Components Components | ADRENODOXIN | ||||||
 Keywords Keywords | ELECTRON TRANSPORT / ELECTRON TRANSPORT PROTEIN / IRON SULFUR PROTEIN / 2FE-2S FERREDOXIN | ||||||
| Function / homology |  Function and homology information Function and homology informationMitochondrial iron-sulfur cluster biogenesis / Pregnenolone biosynthesis / Electron transport from NADPH to Ferredoxin / Endogenous sterols / Protein lipoylation / hormone biosynthetic process / P450-containing electron transport chain / steroid biosynthetic process / cholesterol metabolic process / cellular response to cAMP ...Mitochondrial iron-sulfur cluster biogenesis / Pregnenolone biosynthesis / Electron transport from NADPH to Ferredoxin / Endogenous sterols / Protein lipoylation / hormone biosynthetic process / P450-containing electron transport chain / steroid biosynthetic process / cholesterol metabolic process / cellular response to cAMP / cellular response to forskolin / respiratory electron transport chain / electron transport chain / 2 iron, 2 sulfur cluster binding / electron transfer activity / mitochondrial matrix / protein homodimerization activity / mitochondrion / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Pikuleva, I.A. / Tesh, K. / Waterman, M.R. / Kim, Y. | ||||||
 Citation Citation | Journal: Arch.Biochem.Biophys. / Year: 2000 Title: The tertiary structure of full-length bovine adrenodoxin suggests functional dimers. Authors: Pikuleva, I.A. / Tesh, K. / Waterman, M.R. / Kim, Y. #1: Journal: Structure / Year: 1998Title: New aspects of electron transfer revealed by the crystal structure of a truncated bovine adrenodoxin, Adx(4-108). Authors: Muller, A. / Muller, J.J. / Muller, Y.A. / Uhlmann, H. / Bernhardt, R. / Heinemann, U. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1985 Title: Molecular cloning and amino acid sequence of the precursor form of bovine adrenodoxin: evidence for a previously unidentified COOH-terminal peptide. Authors: Okamura, T. / John, M.E. / Zuber, M.X. / Simpson, E.R. / Waterman, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cje.cif.gz | 97.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cje.ent.gz | 74.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1cje.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cj/1cjeftp://data.pdbj.org/pub/pdb/validation_reports/cj/1cje | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ayfS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 13974.683 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Details: COMPLEXED WITH FE2/S2 (INORGANIC) CLUSTER / Source: (gene. exp.)  #2: Chemical | ChemComp-FES / 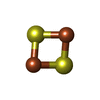  Mass: 175.820 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Fe2S2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 132 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 132 / Source method: isolated from a natural source / Formula: H2OCompound details | THE MATURE ADRENODOXIN IS GENERATED FROM THE ADREDOXIN PRECURSOR (ADX_BOVIN: P00257) BY CLEAVING ...THE MATURE ADRENODOXI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 51 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.5 | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Apr 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→40 Å / Num. obs: 17191 / % possible obs: 92 % / Redundancy: 3.8 % / Biso Wilson estimate: 44.2 Å2 / Rmerge(I) obs: 0.076 / Net I/σ(I): 27 |
| Reflection shell | Resolution: 2.5→2.59 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.29 / Mean I/σ(I) obs: 3.9 / % possible all: 57.4 |
| Reflection | *PLUS Num. obs: 17178 / Num. measured all: 66064 |
| Reflection shell | *PLUS % possible obs: 57 % / Rmerge(I) obs: 0.293 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AYF Resolution: 2.5→40 Å / Rfactor Rfree error: 0.008 / Data cutoff high rms absF: 385760.48 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 59.1 Å2 / ksol: 0.34 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.34 Å / Luzzati d res low obs: 8 Å / Luzzati sigma a obs: 0.33 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.66 Å / Rfactor Rfree error: 0.03 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.4 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 8 Å / Rfactor Rfree: 0.311 / Rfactor Rwork: 0.235 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 44.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
