 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1b3o: TERNARY COMPLEX OF HUMAN TYPE-II INOSINE MONOPHOSPHATE DEHYDROGEN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b3o | ||||||
|---|---|---|---|---|---|---|---|
| Title | TERNARY COMPLEX OF HUMAN TYPE-II INOSINE MONOPHOSPHATE DEHYDROGENASE WITH 6-CL-IMP AND SELENAZOLE ADENINE DINUCLEOTIDE | ||||||
 Components Components | PROTEIN (INOSINE MONOPHOSPHATE DEHYDROGENASE 2) | ||||||
 Keywords Keywords | DEHYDROGENASE / IMPD / IMPDH / GUANINE NUCLEOTIDE SYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationlymphocyte proliferation / 'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / Azathioprine ADME / peroxisomal membrane / GTP biosynthetic process / cellular response to interleukin-4 ...lymphocyte proliferation / 'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / Azathioprine ADME / peroxisomal membrane / GTP biosynthetic process / cellular response to interleukin-4 / circadian rhythm / secretory granule lumen / Potential therapeutics for SARS / ficolin-1-rich granule lumen / nucleotide binding / Neutrophil degranulation / DNA binding / RNA binding / extracellular exosome / extracellular region / membrane / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Colby, T.D. / Vanderveen, K. / Strickler, M.D. / Goldstein, B.M. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1999 Title: Crystal structure of human type II inosine monophosphate dehydrogenase: implications for ligand binding and drug design. Authors: Colby, T.D. / Vanderveen, K. / Strickler, M.D. / Markham, G.D. / Goldstein, B.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b3o.cif.gz | 154.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b3o.ent.gz | 119.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1b3o.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b3/1b3oftp://data.pdbj.org/pub/pdb/validation_reports/b3/1b3o | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.371142, 0.928575, -0.001499), Vector: Details | THE BIOLOGICAL UNIT IS A HOMOTETRAMER. IT IS GENERATED BY A CRYSTALLOGRAPHIC FOURFOLD OPERATION ON MONOMER A (OR MONOMER B), NOT BY A SINGLE OPERATION ON BOTH. | |
-Components
| #1: Protein | Mass: 55874.863 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Variant: TYPE II ISOZYME / Production host:  #2: Chemical | 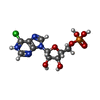  Mass: 367.660 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13ClN4O7P Mass: 367.660 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H13ClN4O7P#3: Chemical |   Mass: 716.348 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H25N7O14P2Se Mass: 716.348 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H25N7O14P2Se#4: Chemical | ChemComp-UNX /   Num. of mol.: 15 / Source method: obtained synthetically Num. of mol.: 15 / Source method: obtained synthetically#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 31 / Source method: isolated from a natural source / Formula: H2OHas protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.9 Å3/Da / Density % sol: 68.93 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: SEE PRIMARY REFERENCE , pH 8.0 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: A1 / Wavelength: 0.91 / Beamline: A1 / Wavelength: 0.91 |
| Detector | Type: CORNELL / Detector: CCD / Date: Aug 1, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→100 Å / Num. obs: 34001 / % possible obs: 86.4 % / Observed criterion σ(I): 0 / Redundancy: 3 % / Biso Wilson estimate: 39.3 Å2 / Rmerge(I) obs: 0.095 / Rsym value: 0.095 / Net I/σ(I): 10 |
| Reflection shell | Resolution: 2.9→3 Å / Redundancy: 1.5 % / Rmerge(I) obs: 0.338 / Mean I/σ(I) obs: 3 / Rsym value: 0.338 / % possible all: 56 |
| Reflection | *PLUS Num. measured all: 126775 |
| Reflection shell | *PLUS % possible obs: 56.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: CORE DOMAIN OF IMPDH MONOMER FROM HAMSTER IMPD/IMP/MPA COMPLEX STRUCTURE Resolution: 2.9→100 Å / Rfactor Rfree error: 0.005 / Data cutoff high rms absF: 182886.58 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: BULK SOLVENT MODEL USED MONOMER B IS THE MORE COMPLETE MONOMER IN THE ASYMMETRIC UNIT, WITH 410 OF 514 RESIDUES FITTED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3 Å / Rfactor Rfree error: 0.028 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.244 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.9 Å / Lowest resolution: 3 Å |
