 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1agm: Refined structure for the complex of acarbose with glucoamylase f... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1agm | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Refined structure for the complex of acarbose with glucoamylase from Aspergillus awamori var. x100 to 2.4 angstroms resolution | |||||||||
 Components Components | GLUCOAMYLASE-471 | |||||||||
 Keywords Keywords | HYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationglucan 1,4-alpha-glucosidase / glucan 1,4-alpha-glucosidase activity / starch binding / fungal-type vacuole / polysaccharide catabolic process Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.3 Å X-RAY DIFFRACTION / Resolution: 2.3 Å | |||||||||
 Authors Authors | Aleshin, A.E. / Firsov, L.M. / Honzatko, R.B. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1994 Title: Refined structure for the complex of acarbose with glucoamylase from Aspergillus awamori var. X100 to 2.4-A resolution. Authors: Aleshin, A.E. / Firsov, L.M. / Honzatko, R.B. #1: Journal: J.Mol.Biol. / Year: 1994Title: Refined Crystal Structures of Glucoamylase from Aspergillus Awamori Var. X100 Authors: Aleshin, A.E. / Hoffman, C. / Firsov, L.M. / Honzatko, R.B. #2: Journal: Biochemistry / Year: 1993Title: Refined Structure of the Complex of 1-Deoxynojirimycin with Glucoamylase from Aspergillus Awamori Var. X100 to 2.4 Angstroms Resolution Authors: Harris, E.M.S. / Aleshin, A.E. / Firsov, L.M. / Honzatko, R.B. #3: Journal: J.Biol.Chem. / Year: 1992Title: Crystal Structure of Glucoamylase from Aspergillus Awamori Var. X100 to 2.2 Angstroms Resolution Authors: Aleshin, A.E. / Golubev, A. / Firsov, L.M. / Honzatko, R.B. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX THE POLYPEPTIDE CHAIN FOLDS INTO AN ALPHA/ALPHA-BARREL, COMPRISING 12 HELICES. | |||||||||
| Remark 700 | SHEET MOST OF THE SHEETS FOR GLUCOAMYLASE-471 ARE HAIRPIN LOOPS THAT CONNECT HELICES. THESE LOOPS ...SHEET MOST OF THE SHEETS FOR GLUCOAMYLASE-471 ARE HAIRPIN LOOPS THAT CONNECT HELICES. THESE LOOPS HAVE TWO OR MORE H-BONDS BETWEEN THE ANTIPARALLEL STRANDS THAT COMPRISE THEM. IN ADDITION INDIVIDUAL LOOPS PACK TOGETHER, BUT THERE EXISTS GENERALLY ONLY ONE H-BOND BETWEEN A LOOP AND ITS NEIGHBOR. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1agm.cif.gz | 126.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1agm.ent.gz | 94.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1agm.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ag/1agmftp://data.pdbj.org/pub/pdb/validation_reports/ag/1agm | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUES GLY 23 AND ALA 24 FORM A CIS PEPTIDE. / 2: CIS PROLINE - PRO 46 / 3: CIS PROLINE - PRO 123 4: RESIDUES ASN 171 AND ASN 395 ARE SITES OF N-GLYCOSYLATION. 5: RESIDUES SER 443, SER 444, THR 452, SER 453, SER 455, THR 457, SER 459, SER 460, THR 462 AND THR 464 ARE SITES OF O-GLYCOSYLATION. |
-Components
-Protein / Non-polymers , 2 types, 536 molecules A
| #1: Protein | Mass: 50450.730 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P23176, UniProt: P69327*PLUS, glucan 1,4-alpha-glucosidase |
|---|---|
| #6: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 535 / Source method: isolated from a natural source / Formula: H2O |
-Sugars , 4 types, 14 molecules 
| #2: Polysaccharide | alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-3)-beta-D-mannopyranose-(1-4)-2-acetamido-2- ...alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-3)-beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source | ||
|---|---|---|---|
| #3: Polysaccharide | alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-3)-[alpha-D- ...alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-2)-alpha-D-mannopyranose-(1-3)-[alpha-D-mannopyranose-(1-3)-alpha-D-mannopyranose-(1-6)]beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source | ||
| #4: Polysaccharide | 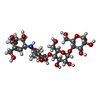  Type: oligosaccharide, Oligosaccharide / Class: Inhibitor / Mass: 645.606 Da / Num. of mol.: 2 Type: oligosaccharide, Oligosaccharide / Class: Inhibitor / Mass: 645.606 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-acarbose #5: Sugar | ChemComp-MAN /  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 10 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 10Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57.64 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 5.95 / Method: vapor diffusion | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.3 Å / Num. all: 26797 / Num. obs: 21212 / Num. measured all: 51528 / Rmerge(I) obs: 0.032 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.3→10 Å / σ(F): 1 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.124 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
