 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1a0j | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF A NON-PSYCHROPHILIC TRYPSIN FROM A COLD-ADAPTED FISH SPECIES. | ||||||
 要素 要素 | TRYPSIN | ||||||
 キーワード キーワード | SERINE PROTEASE / SERINE PROTEINASE / TRYPSIN / HYDROLASE | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報trypsin / digestion / serine-type endopeptidase activity / proteolysis / : / metal ion binding 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å | ||||||
 データ登録者 データ登録者 | Schroeder, H.-K. / Willassen, N.P. / Smalaas, A.O. | ||||||
 引用 引用 | ジャーナル: Acta Crystallogr.,Sect.D / 年: 1998 タイトル: Structure of a non-psychrophilic trypsin from a cold-adapted fish species. 著者: Schroder, H.K. / Willassen, N.P. / Smalas, A.O. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1a0j.cif.gz | 188.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1a0j.ent.gz | 148.4 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1a0j.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/a0/1a0jftp://data.pdbj.org/pub/pdb/validation_reports/a0/1a0j | HTTPS FTP |
|---|
-関連構造データ












-リンク
 PDBj
PDBj


- 集合体
集合体
| 登録構造単位 | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||
| 単位格子 |
| ||||||||||||||||||||||||||||
| 非結晶学的対称性 (NCS) | NCS oper:
|
-要素
| #1: タンパク質 | 分子量: 23803.662 Da / 分子数: 4 / 由来タイプ: 天然 詳細: BENZAMIDINE INHIBITOR IN THE ACTIVE SITE. THE AMINO ACID NUMBERING SCHEME USED IS ADOPTED FROM CHYMOTRYPSINOGEN. 由来: (天然) #2: 化合物 | ChemComp-CA / |   分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca 分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca#3: 化合物 | ChemComp-SO4 /   分子量: 96.063 Da / 分子数: 10 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 10 / 由来タイプ: 合成 / 式: SO4#4: 化合物 | ChemComp-BEN / 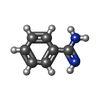  分子量: 120.152 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C7H8N2 分子量: 120.152 Da / 分子数: 4 / 由来タイプ: 合成 / 式: C7H8N2#5: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 379 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 379 / 由来タイプ: 天然 / 式: H2OHas protein modification | Y | 配列の詳細 | THE AMINO ACID NUMBERING SCHEME USED IS ADOPTED FROM CHYMOTRYPS | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 4 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.2 Å3/Da / 溶媒含有率: 44 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | pH: 4.6 / 詳細: pH 4.6 | |||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 310 K / 手法: 蒸気拡散法詳細: drop contained 1:1 mixture of protein and reservoir solution | |||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 295 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ESRF  / ビームライン: BM1A / 波長: 0.873 / ビームライン: BM1A / 波長: 0.873 |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 1996年8月1日 |
| 放射 | 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.873 Å / 相対比: 1 |
| 反射 | 解像度: 1.7→28 Å / Num. obs: 76357 / % possible obs: 81.2 % / Observed criterion σ(I): 1 / 冗長度: 5.9 % / Biso Wilson estimate: 19.6 Å2 / Rmerge(I) obs: 0.123 / Net I/σ(I): 3.6 |
| 反射 シェル | 解像度: 1.7→1.79 Å / 冗長度: 2 % / Rmerge(I) obs: 0.258 / Mean I/σ(I) obs: 2.9 / % possible all: 52.3 |
| 反射 | *PLUS Num. measured all: 451079 |
| 反射 シェル | *PLUS % possible obs: 52.3 % |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: PDB ENTRY 3PTB BOVINE TRYPSIN, WITH CORRECT AMINO ACID SEQUENCE SUPERIMPOSED ONTO ANIONIC SALMON TRYPSIN 2TBS. 解像度: 1.7→8 Å / 交差検証法: THROUGHOUT / σ(F): 3 詳細: NCS RESTRAINTS WERE INCLUDED IN THE FIRST REFINEMENT ROUNDS, THEN LEFT OUT. THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A). THE ELECTRON DENSITY ...詳細: NCS RESTRAINTS WERE INCLUDED IN THE FIRST REFINEMENT ROUNDS, THEN LEFT OUT. THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A). THE ELECTRON DENSITY INDICATES A CALCIUM ION FOR ONLY ONE OF THE FOUR MOLECULES (MOL A).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 22.93 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.18 Å / Luzzati sigma a free: 0.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.7→1.76 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3.8 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | *PLUS Rfactor obs: 0.2347 |
