 ムービー
ムービー コントローラー
コントローラー

[日本語] English
 万見
万見- EMDB-9202: T20S proteasome using with published picks (EMPIAR-10025 reprocessing) -
+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-9202 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | T20S proteasome using with published picks (EMPIAR-10025 reprocessing) | |||||||||
 マップデータ マップデータ | CryoSparc v0 sharpened map from published picks. | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |   Thermoplasma acidophilum (好酸性) Thermoplasma acidophilum (好酸性) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.99 Å | |||||||||
 データ登録者 データ登録者 | Bepler T / Morin A / Brasch J / Shapiro L / Noble AJ / Berger B | |||||||||
 引用 引用 | ジャーナル: Elife / 年: 2015 タイトル: 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. 著者: Melody G Campbell / David Veesler / Anchi Cheng / Clinton S Potter / Bridget Carragher /  要旨: Recent developments in detector hardware and image-processing software have revolutionized single particle cryo-electron microscopy (cryoEM) and led to a wave of near-atomic resolution (typically ...Recent developments in detector hardware and image-processing software have revolutionized single particle cryo-electron microscopy (cryoEM) and led to a wave of near-atomic resolution (typically ∼3.3 Å) reconstructions. Reaching resolutions higher than 3 Å is a prerequisite for structure-based drug design and for cryoEM to become widely interesting to pharmaceutical industries. We report here the structure of the 700 kDa Thermoplasma acidophilum 20S proteasome (T20S), determined at 2.8 Å resolution by single-particle cryoEM. The quality of the reconstruction enables identifying the rotameric conformation adopted by some amino-acid side chains (rotamers) and resolving ordered water molecules, in agreement with the expectations for crystal structures at similar resolutions. The results described in this manuscript demonstrate that single particle cryoEM is capable of competing with X-ray crystallography for determination of protein structures of suitable quality for rational drug design. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_9202.map.gz | 229.7 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-9202-v30.xmlemd-9202.xml | 18.4 KB 18.4 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_9202.png emd_9202.png | 175.8 KB | ||
| マスクデータ | emd_9202_msk_1.map | 244.1 MB | マスクマップ | |
| その他 | emd_9202_additional.map.gzemd_9202_half_map_1.map.gzemd_9202_half_map_2.map.gz | 120.6 MB 226.3 MB 226.3 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-9202ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9202 http://ftp.pdbj.org/pub/emdb/structures/EMD-9202ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9202 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_9202_validation.pdf.gz | 77.8 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_9202_full_validation.pdf.gz | 76.9 KB | 表示 | |
| XML形式データ | emd_9202_validation.xml.gz | 491 B | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-9202ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-9202 | HTTPS FTP |
-関連構造データ














-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_9202.map.gz / 形式: CCP4 / 大きさ: 244.1 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | CryoSparc v0 sharpened map from published picks. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
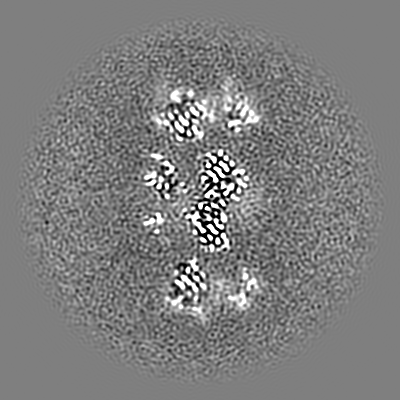
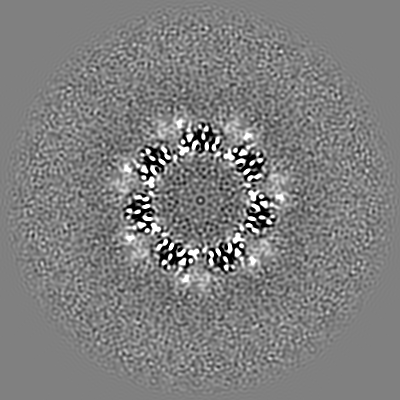
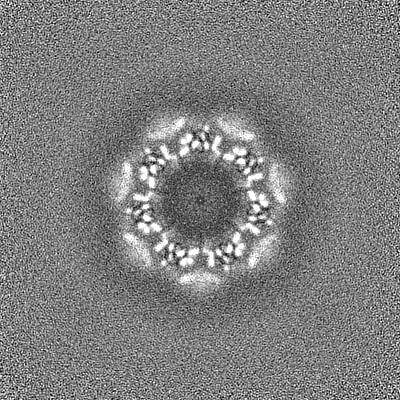
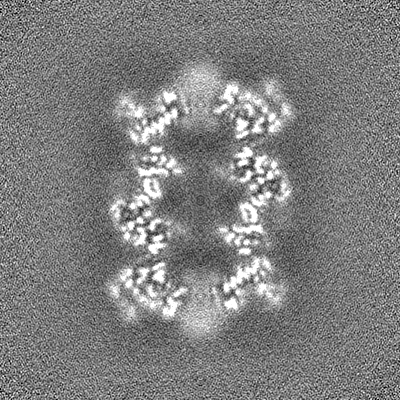
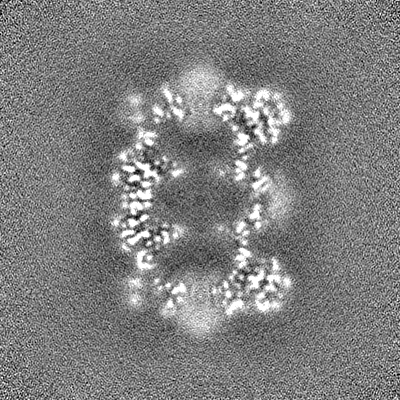
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.6575 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
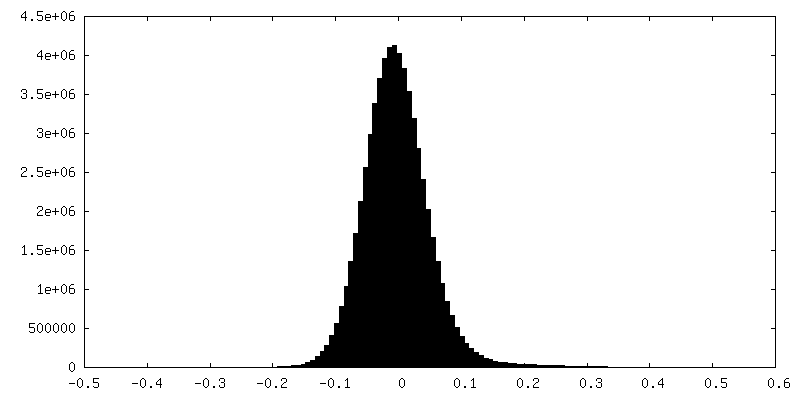
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-添付データ
-マスク #1
| ファイル | emd_9202_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
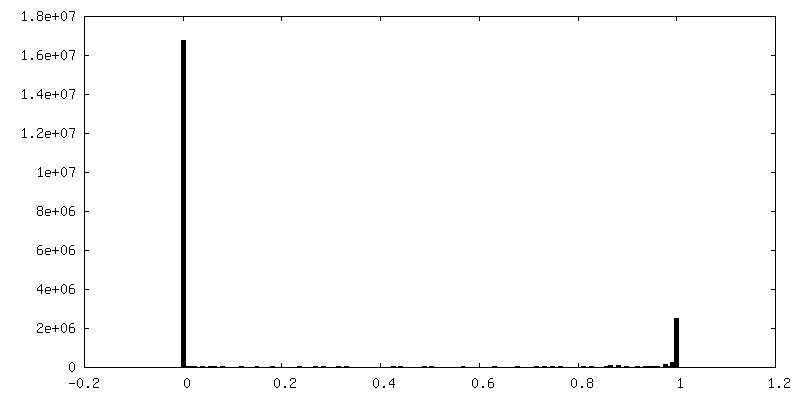
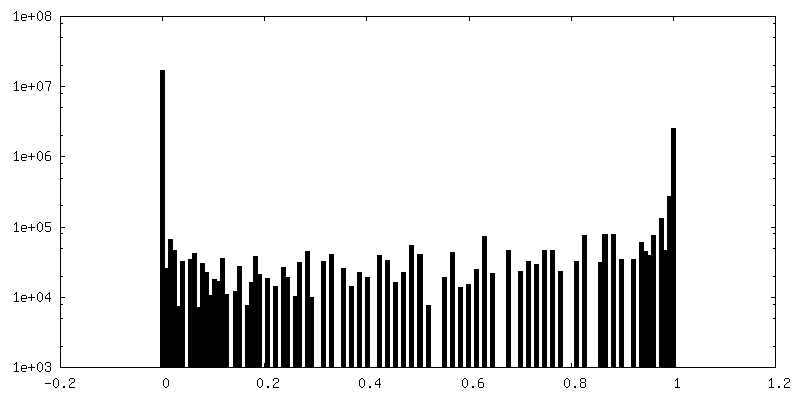
| 密度ヒストグラム |






-追加マップ: CryoSparc v0 unsharpened map from published picks.
| ファイル | emd_9202_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | CryoSparc v0 unsharpened map from published picks. | ||||||||||||
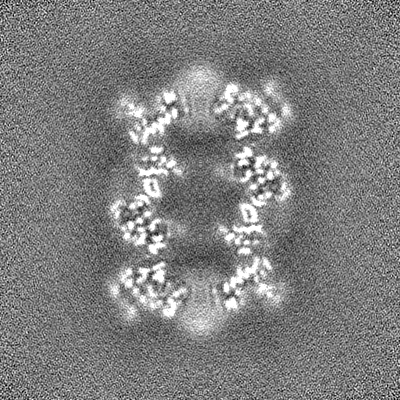
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




-ハーフマップ: CryoSparc v0 halfmap from published picks.
| ファイル | emd_9202_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | CryoSparc v0 halfmap from published picks. | ||||||||||||
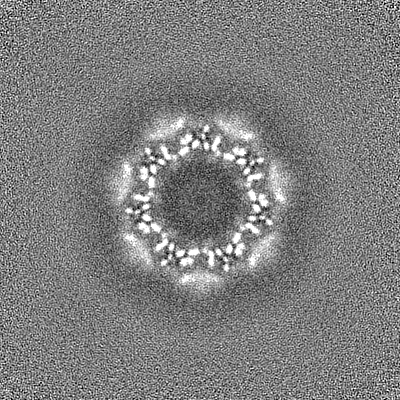
| 投影像・断面図 |
| ||||||||||||
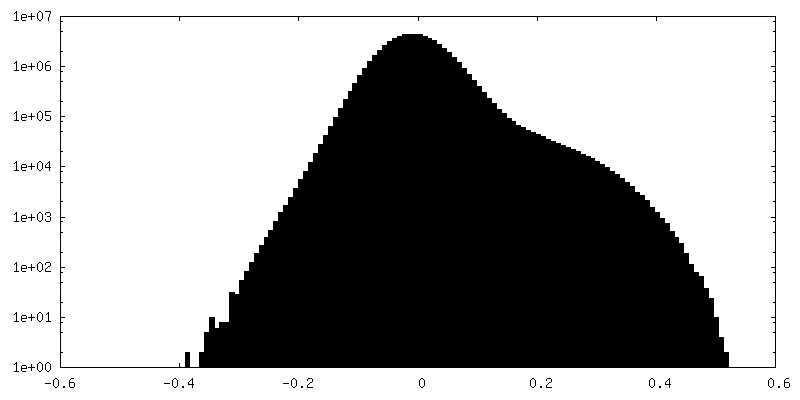
| 密度ヒストグラム |




-ハーフマップ: CryoSparc v0 halfmap from published picks.
| ファイル | emd_9202_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | CryoSparc v0 halfmap from published picks. | ||||||||||||
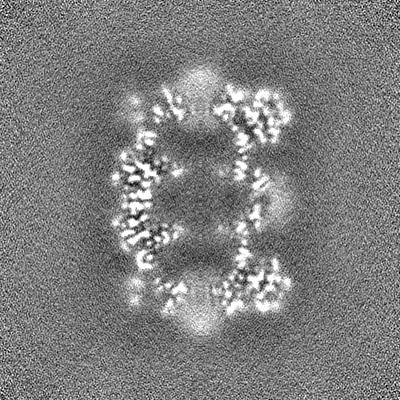
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |



- 試料の構成要素
試料の構成要素
-全体 : T20S proteasome
| 全体 | 名称: T20S proteasome |
|---|---|
| 要素 |
|
-超分子 #1: T20S proteasome
| 超分子 | 名称: T20S proteasome / タイプ: complex / ID: 1 / 親要素: 0 |
|---|---|
| 由来(天然) | 生物種: Thermoplasma acidophilum (好酸性) |
| 組換発現 | 生物種: Thermoplasma acidophilum (好酸性) |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.21 mg/mL |
|---|---|
| 緩衝液 | pH: 7.8 / 詳細: 20 mM Tris, 150 mM NaCl |
| グリッド | 詳細: unspecified |
| 凍結 | 凍結剤: ETHANE / 装置: GATAN CRYOPLUNGE 3 詳細: Blot for 2.5 seconds before plunging. Vitrification carried out at room temperature.. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: SUPER-RESOLUTION / 平均電子線量: 53.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): 2.4 µm / 最小 デフォーカス(公称値): 0.9 µm / 倍率(公称値): 22500 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
