 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-9202: T20S proteasome using with published picks (EMPIAR-10025 reprocessing) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-9202 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | T20S proteasome using with published picks (EMPIAR-10025 reprocessing) | |||||||||
 Map data Map data | CryoSparc v0 sharpened map from published picks. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Thermoplasma acidophilum (acidophilic) Thermoplasma acidophilum (acidophilic) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.99 Å | |||||||||
 Authors Authors | Bepler T / Morin A / Brasch J / Shapiro L / Noble AJ / Berger B | |||||||||
 Citation Citation | Journal: Elife / Year: 2015 Title: 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. Authors: Melody G Campbell / David Veesler / Anchi Cheng / Clinton S Potter / Bridget Carragher /  Abstract: Recent developments in detector hardware and image-processing software have revolutionized single particle cryo-electron microscopy (cryoEM) and led to a wave of near-atomic resolution (typically ...Recent developments in detector hardware and image-processing software have revolutionized single particle cryo-electron microscopy (cryoEM) and led to a wave of near-atomic resolution (typically ∼3.3 Å) reconstructions. Reaching resolutions higher than 3 Å is a prerequisite for structure-based drug design and for cryoEM to become widely interesting to pharmaceutical industries. We report here the structure of the 700 kDa Thermoplasma acidophilum 20S proteasome (T20S), determined at 2.8 Å resolution by single-particle cryoEM. The quality of the reconstruction enables identifying the rotameric conformation adopted by some amino-acid side chains (rotamers) and resolving ordered water molecules, in agreement with the expectations for crystal structures at similar resolutions. The results described in this manuscript demonstrate that single particle cryoEM is capable of competing with X-ray crystallography for determination of protein structures of suitable quality for rational drug design. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_9202.map.gz | 229.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-9202-v30.xmlemd-9202.xml | 18.4 KB 18.4 KB | Display Display | EMDB header |
| Images |  emd_9202.png emd_9202.png | 175.8 KB | ||
| Masks | emd_9202_msk_1.map | 244.1 MB | Mask map | |
| Others | emd_9202_additional.map.gzemd_9202_half_map_1.map.gzemd_9202_half_map_2.map.gz | 120.6 MB 226.3 MB 226.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-9202ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9202 http://ftp.pdbj.org/pub/emdb/structures/EMD-9202ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9202 | HTTPS FTP |
-Related structure data
| Related structure data |  9194C  9201C  9206C  9207C  9208C  9209C  9210C  9211C C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_9202.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoSparc v0 sharpened map from published picks. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
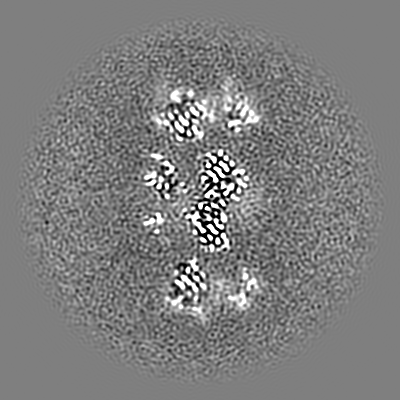
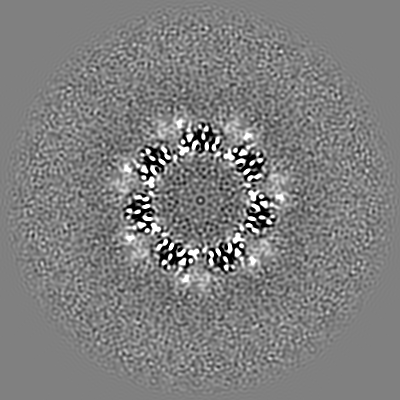
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.6575 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Mask #1
| File | emd_9202_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
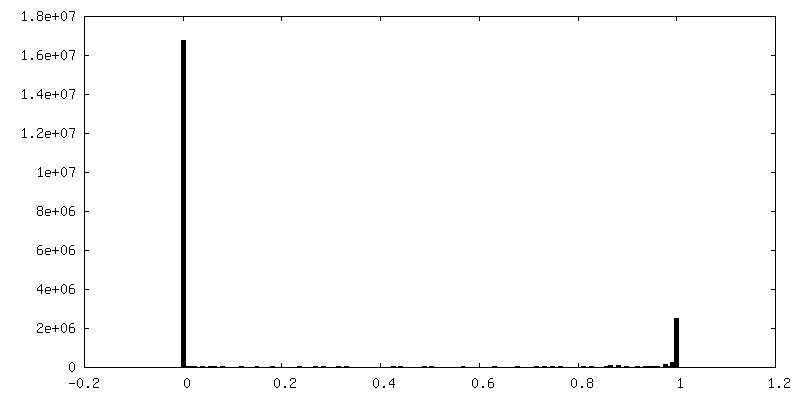
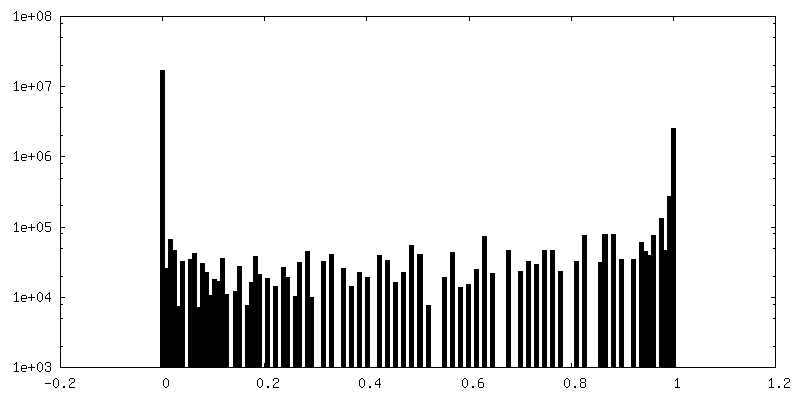
| Density Histograms |






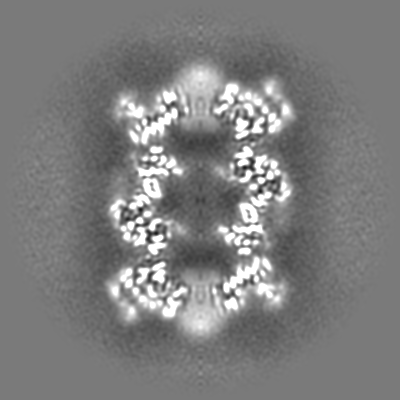
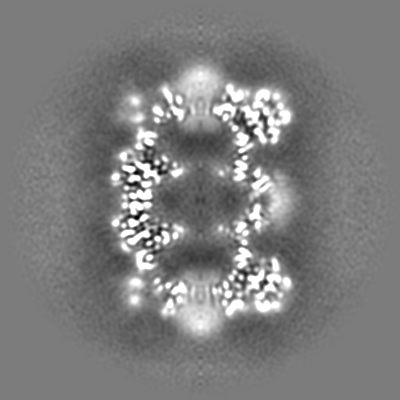
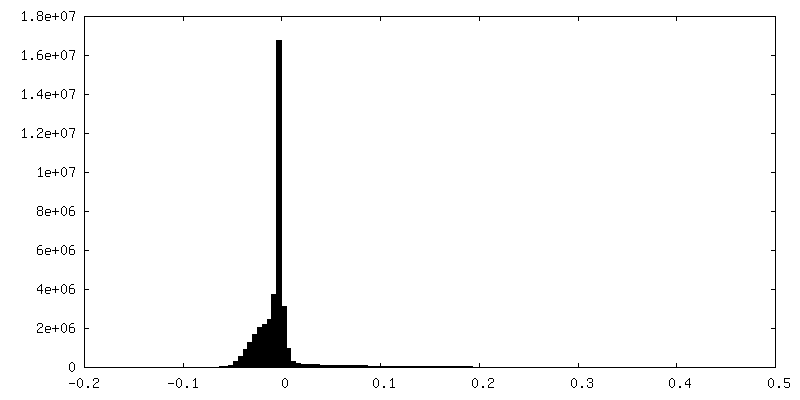
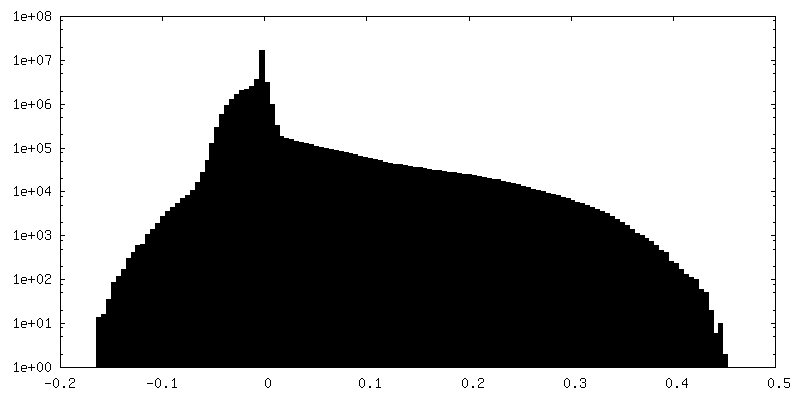
-Additional map: CryoSparc v0 unsharpened map from published picks.
| File | emd_9202_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoSparc v0 unsharpened map from published picks. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: CryoSparc v0 halfmap from published picks.
| File | emd_9202_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoSparc v0 halfmap from published picks. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



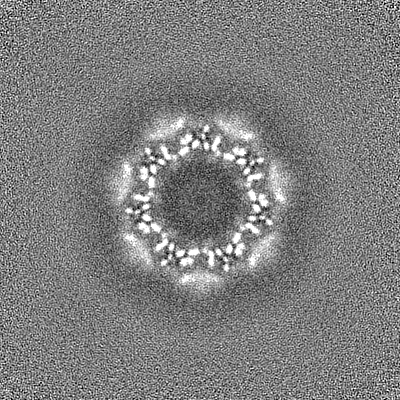
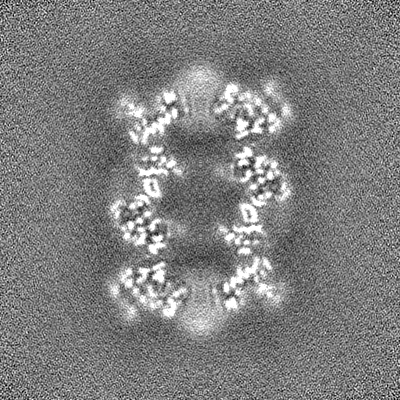
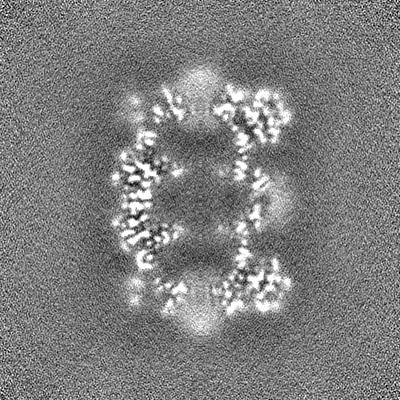

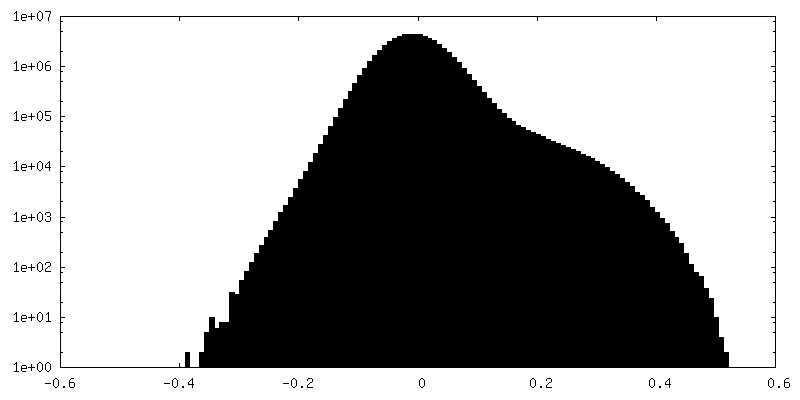
-Half map: CryoSparc v0 halfmap from published picks.
| File | emd_9202_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoSparc v0 halfmap from published picks. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



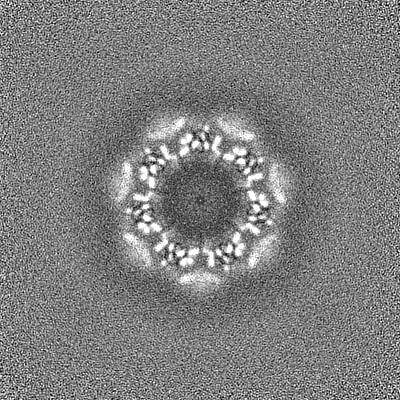
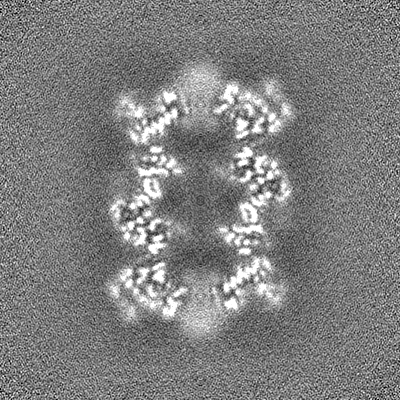
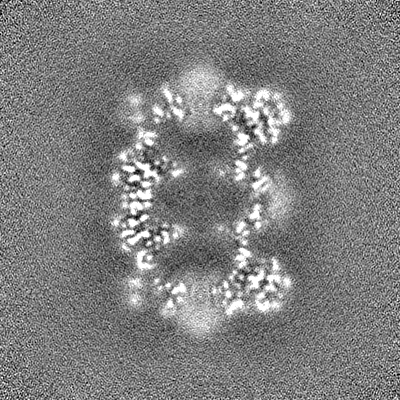
- Sample components
Sample components
-Entire : T20S proteasome
| Entire | Name: T20S proteasome |
|---|---|
| Components |
|
-Supramolecule #1: T20S proteasome
| Supramolecule | Name: T20S proteasome / type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Thermoplasma acidophilum (acidophilic) |
| Recombinant expression | Organism: Thermoplasma acidophilum (acidophilic) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.21 mg/mL |
|---|---|
| Buffer | pH: 7.8 / Details: 20 mM Tris, 150 mM NaCl |
| Grid | Details: unspecified |
| Vitrification | Cryogen name: ETHANE / Instrument: GATAN CRYOPLUNGE 3 Details: Blot for 2.5 seconds before plunging. Vitrification carried out at room temperature.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Average electron dose: 53.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.4 µm / Nominal defocus min: 0.9 µm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
