 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-9012: Sub-2 Angstrom Ewald Curvature Corrected Single-Particle Cryo-EM ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-9012 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Sub-2 Angstrom Ewald Curvature Corrected Single-Particle Cryo-EM Reconstruction of AAV-2 L336C | ||||||||||||
 Map data Map data | Final map reconstructed with a box of 800, clipped to 360, and then resampled to 720 pixels | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | AAV-2 L336C / Gene therapy / Ewald sphere curvature correction / Per particle CTF / VIRUS LIKE PARTICLE | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont entry into host cell via permeabilization of host membrane / host cell nucleolus / T=1 icosahedral viral capsid / clathrin-dependent endocytosis of virus by host cell / virion attachment to host cell / structural molecule activity Similarity search - Function | ||||||||||||
| Biological species |  Adeno-associated virus 2 Srivastava/1982 / Adeno-associated virus - 2 Adeno-associated virus 2 Srivastava/1982 / Adeno-associated virus - 2 | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 1.86 Å | ||||||||||||
 Authors Authors | Tan YZ / Aiyer S | ||||||||||||
| Funding support |  United States, 3 items United States, 3 items
| ||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2018 Title: Sub-2 Å Ewald curvature corrected structure of an AAV2 capsid variant. Authors: Yong Zi Tan / Sriram Aiyer / Mario Mietzsch / Joshua A Hull / Robert McKenna / Joshua Grieger / R Jude Samulski / Timothy S Baker / Mavis Agbandje-McKenna / Dmitry Lyumkis / Abstract: Single-particle cryogenic electron microscopy (cryo-EM) provides a powerful methodology for structural biologists, but the resolutions typically attained with experimentally determined structures ...Single-particle cryogenic electron microscopy (cryo-EM) provides a powerful methodology for structural biologists, but the resolutions typically attained with experimentally determined structures have lagged behind microscope capabilities. Here, we exploit several technical advances to improve resolution, including per-particle contrast transfer function (CTF) refinement and correction for Ewald sphere curvature. The latter is demonstrated with several experimental samples and should become more standard as resolutions increase or at lower microscope accelerating voltages. The combined application of the described methods to micrographs recorded on a Titan Krios enables structure determination at ~1.86-Å resolution of an adeno-associated virus serotype 2 variant (AAV2), an important gene-delivery vehicle. The resulting structural details provide an improved model for understanding the biology of AAV that will guide future vector development for gene therapy. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_9012.map.gz | 1.3 GB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-9012-v30.xmlemd-9012.xml | 23.3 KB 23.3 KB | Display Display | EMDB header |
| Images |  emd_9012.png emd_9012.png | 86.1 KB | ||
| Filedesc metadata | emd-9012.cif.gz | 7.4 KB | ||
| Others | emd_9012_additional.map.gzemd_9012_half_map_1.map.gzemd_9012_half_map_2.map.gz | 47.3 MB 94 MB 94 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-9012ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9012 http://ftp.pdbj.org/pub/emdb/structures/EMD-9012ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9012 | HTTPS FTP |
-Related structure data
| Related structure data |  6e9dMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10202 (Title: Sub-2 Å Single-Particle Cryo-EM Reconstruction of AAV2-L336C Data size: 5.7 TB Data #1: Unaligned, compressed, raw movie frames of AAV2-L336C [micrographs - multiframe] Data #2: MotionCor2 aligned, gain corrected, magnification anisotropy corrected, Fourier bin by 2 movie frames of AAV2-L336C [micrographs - multiframe] Data #3: MotionCor2 aligned, gain corrected, magnification anisotropy corrected, Fourier bin by 2, dose weighted, summed micrographs of AAV2-L336C [micrographs - single frame] Data #4: MotionCor2 aligned, gain corrected, magnification anisotropy corrected, Fourier bin by 2, not dose weighted, summed micrographs of AAV2-L336C [micrographs - single frame] Data #5: MotionCor2 aligned, gain corrected, magnification anisotropy corrected, Fourier bin by 2, not dose weighted, frames 5 to 19 summed micrographs of AAV2-L336C [micrographs - single frame] Data #6: Final particle stack of AAV2-L336C using frames 5 to 19 [micrographs - single frame] Data #7: Final particle stack of AAV2-L336C grouped into frames 5-9, 10-14 and 15-19, after rotational alignment [micrographs - single frame]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_9012.map.gz / Format: CCP4 / Size: 1.4 GB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Final map reconstructed with a box of 800, clipped to 360, and then resampled to 720 pixels | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.394 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















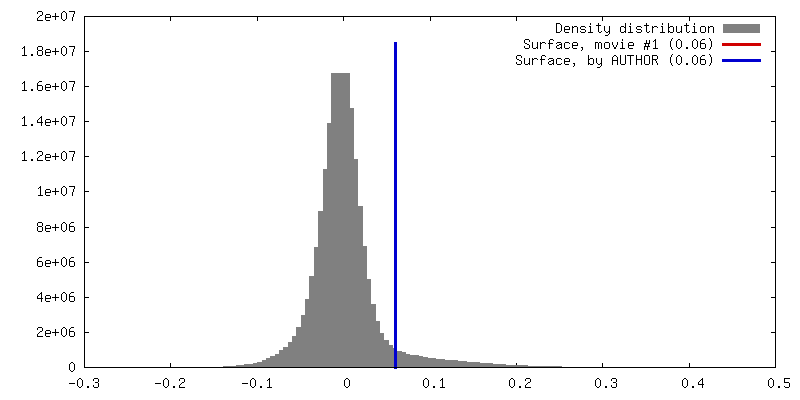

-Supplemental data
-Additional map: Raw 3D FSC volume generated from the half maps
| File | emd_9012_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Raw 3D FSC volume generated from the half maps | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







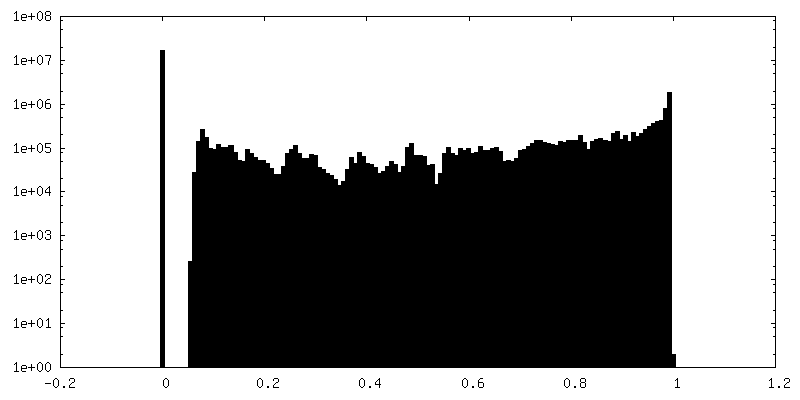
-Half map: Second half map reconstructed with a box of...
| File | emd_9012_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Second half map reconstructed with a box of 800, clipped to 360 pixels | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






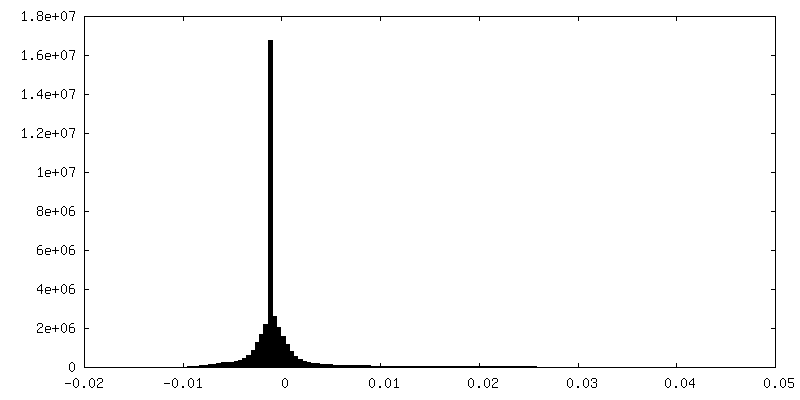
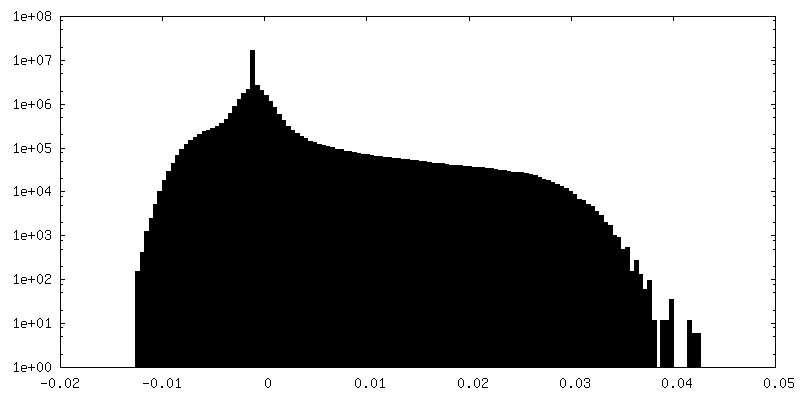
-Half map: First half map reconstructed with a box of 800, clipped to 360 pixels
| File | emd_9012_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | First half map reconstructed with a box of 800, clipped to 360 pixels | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : Adeno-associated virus - 2
| Entire | Name: Adeno-associated virus - 2 |
|---|---|
| Components |
|
-Supramolecule #1: Adeno-associated virus - 2
| Supramolecule | Name: Adeno-associated virus - 2 / type: virus / ID: 1 / Parent: 0 / Macromolecule list: #1 / NCBI-ID: 10804 / Sci species name: Adeno-associated virus - 2 / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: STRAIN / Virus enveloped: No / Virus empty: Yes |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Molecular weight | Theoretical: 3.9 MDa |
| Virus shell | Shell ID: 1 / Name: Icosahedral capsid / Diameter: 250.0 Å / T number (triangulation number): 1 |
-Macromolecule #1: Capsid protein VP1
| Macromolecule | Name: Capsid protein VP1 / type: protein_or_peptide / ID: 1 / Number of copies: 60 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Adeno-associated virus 2 Srivastava/1982 |
| Molecular weight | Theoretical: 82.021336 KDa |
| Recombinant expression | Organism:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm) |
| Sequence | String: MAADGYLPDW LEDTLSEGIR QWWKLKPGPP PPKPAERHKD DSRGLVLPGY KYLGPFNGLD KGEPVNEADA AALEHDKAYD RQLDSGDNP YLKYNHADAE FQERLKEDTS FGGNLGRAVF QAKKRVLEPL GLVEEPVKTA PGKKRPVEHS PVEPDSSSGT G KAGQQPAR ...String: MAADGYLPDW LEDTLSEGIR QWWKLKPGPP PPKPAERHKD DSRGLVLPGY KYLGPFNGLD KGEPVNEADA AALEHDKAYD RQLDSGDNP YLKYNHADAE FQERLKEDTS FGGNLGRAVF QAKKRVLEPL GLVEEPVKTA PGKKRPVEHS PVEPDSSSGT G KAGQQPAR KRLNFGQTGD ADSVPDPQPL GQPPAAPSGL GTNTMATGSG APMADNNEGA DGVGNSSGNW HCDSTWMGDR VI TTSTRTW ALPTYNNHLY KQISSQSGAS NDNHYFGYST PWGYFDFNRF HCHFSPRDWQ RLINNNWGFR PKRLNFKLFN IQV KEVTQN DGTTTIANNC TSTVQVFTDS EYQLPYVLGS AHQGCLPPFP ADVFMVPQYG YLTLNNGSQA VGRSSFYCLE YFPS QMLRT GNNFTFSYTF EDVPFHSSYA HSQSLDRLMN PLIDQYLYYL SRTNTPSGTT TQSRLQFSQA GASDIRDQSR NWLPG PCYR QQRVSKTSAD NNNSEYSWTG ATKYHLNGRD SLVNPGPAMA SHKDDEEKFF PQSGVLIFGK QGSEKTNVDI EKVMIT DEE EIRTTNPVAT EQYGSVSTNL QRGNRQAATA DVNTQGVLPG MVWQDRDVYL QGPIWAKIPH TDGHFHPSPL MGGFGLK HP PPQILIKNTP VPANPSTTFS AAKFASFITQ YSTGQVSVEI EWELQKENSK RWNPEIQYTS NYNKSVNVDF TVDTNGVY S EPRPIGTRYL TRNL UniProtKB: Capsid protein VP1 |
-Macromolecule #2: water
| Macromolecule | Name: water / type: ligand / ID: 2 / Number of copies: 11820 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.7 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
| ||||||||||||
| Grid | Model: Quantifoil, UltrAuFoil, R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - topology: HOLEY / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Time: 5 sec. / Pretreatment - Atmosphere: OTHER | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 80 % / Chamber temperature: 277 K / Instrument: HOMEMADE PLUNGER Details: Double blotting was used to increase particle concentration. Sample was added to a plasma-cleaned (Gatan Solarus) grid and blotted away using Whatman grade 4 filter paper after 20 second wait time.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Details | Beam tilt in 8 directions +/- 5 mrad |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 5-19 / Number grids imaged: 1 / Number real images: 1317 / Average exposure time: 3.5 sec. / Average electron dose: 22.5 e/Å2 / Details: Stage shift mode for data collection |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 0.6 µm / Nominal magnification: 37000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Overall B value: 61.9 / Target criteria: Correlation coefficient, EM ringer score |
| Output model | PDB-6e9d: |

