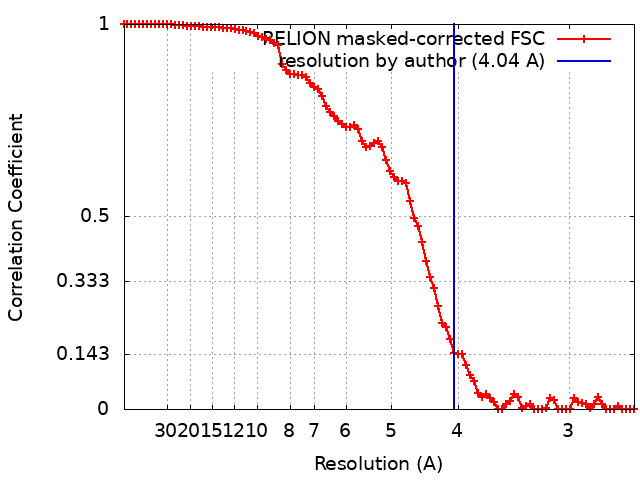
ジャーナル: Nature / 年: 2016 タイトル: Pre-fusion structure of a human coronavirus spike protein. 著者: Robert N Kirchdoerfer / Christopher A Cottrell / Nianshuang Wang / Jesper Pallesen / Hadi M Yassine / Hannah L Turner / Kizzmekia S Corbett / Barney S Graham / Jason S McLellan / Andrew B Ward / 要旨: HKU1 is a human betacoronavirus that causes mild yet prevalent respiratory disease, and is related to the zoonotic SARS and MERS betacoronaviruses, which have high fatality rates and pandemic ...HKU1 is a human betacoronavirus that causes mild yet prevalent respiratory disease, and is related to the zoonotic SARS and MERS betacoronaviruses, which have high fatality rates and pandemic potential. Cell tropism and host range is determined in part by the coronavirus spike (S) protein, which binds cellular receptors and mediates membrane fusion. As the largest known class I fusion protein, its size and extensive glycosylation have hindered structural studies of the full ectodomain, thus preventing a molecular understanding of its function and limiting development of effective interventions. Here we present the 4.0 Å resolution structure of the trimeric HKU1 S protein determined using single-particle cryo-electron microscopy. In the pre-fusion conformation, the receptor-binding subunits, S1, rest above the fusion-mediating subunits, S2, preventing their conformational rearrangement. Surprisingly, the S1 C-terminal domains are interdigitated and form extensive quaternary interactions that occlude surfaces known in other coronaviruses to bind protein receptors. These features, along with the location of the two protease sites known to be important for coronavirus entry, provide a structural basis to support a model of membrane fusion mediated by progressive S protein destabilization through receptor binding and proteolytic cleavage. These studies should also serve as a foundation for the structure-based design of betacoronavirus vaccine immunogens.
タイプ: NEGATIVE / 材質: uranyl formate 詳細: 3 uL sample was applied to grid for 30 seconds and then blotted. Grids were stained with 3 uL 1% uranyl formate for 60 seconds followed by blotting.
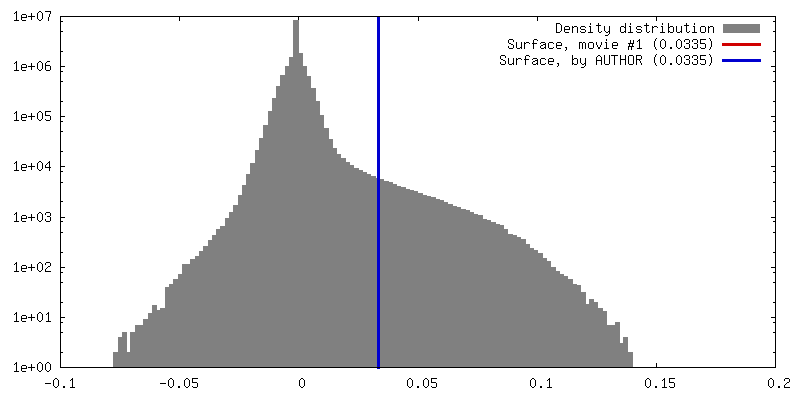
選択した数: 39164 詳細: 2188 particles were selected from a subset of the data using DoG Picker. These particles were used to generate a 3D model from which back projections were derived. Back projection images were ...詳細: 2188 particles were selected from a subset of the data using DoG Picker. These particles were used to generate a 3D model from which back projections were derived. Back projection images were used as templates for picking particles from the entire dataset using FindEM.
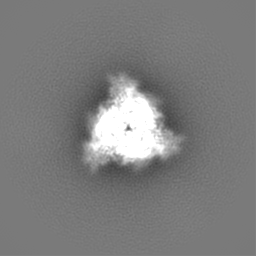
初期モデル
モデルのタイプ: INSILICO MODEL In silico モデル: Initial model was generated with EMAN2 using a selection of 2D classifications from a subset of the data containing 2188 particles.
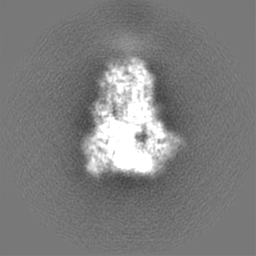
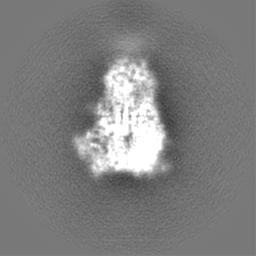
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Human coronavirus HKU1 (isolate N5) (ウイルス) /
Human coronavirus HKU1 (isolate N5) (ウイルス) /  データ登録者
データ登録者 引用
引用
 構造の表示
構造の表示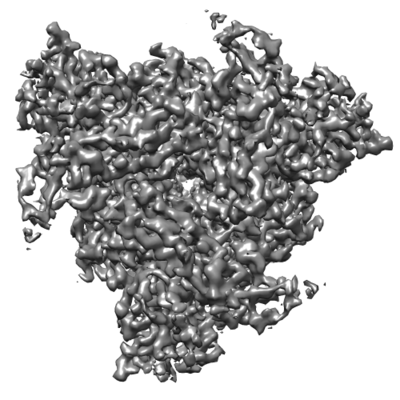
 ダウンロードとリンク
ダウンロードとリンク emd_8069.png
emd_8069.png http://ftp.pdbj.org/pub/emdb/structures/EMD-8069
http://ftp.pdbj.org/pub/emdb/structures/EMD-8069













 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















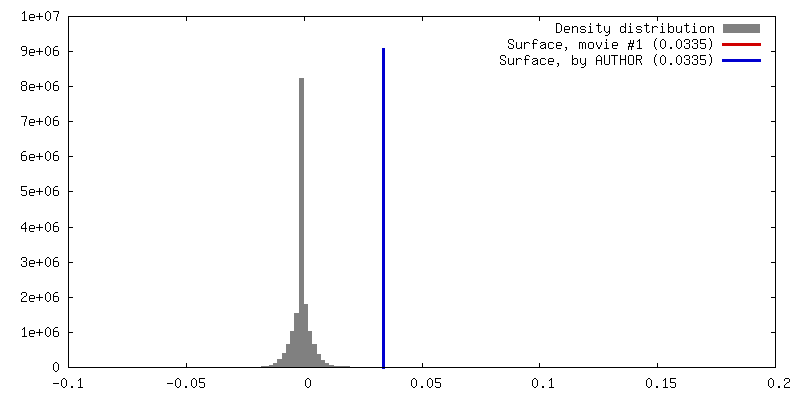


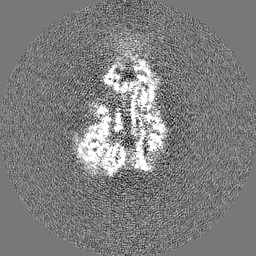
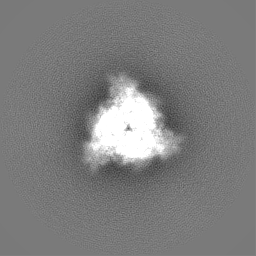
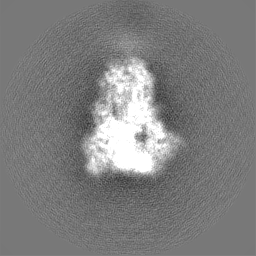
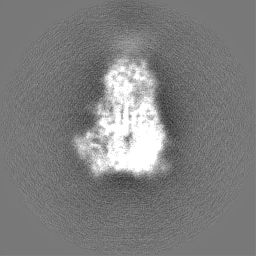


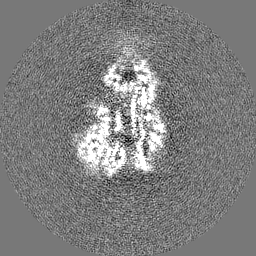
 試料の構成要素
試料の構成要素 Homo sapiens (ヒト)
Homo sapiens (ヒト) 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN