 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-7952 | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Caseinolytic protease (ClpP) from Staphylococcus aureus mutant - V7A | |||||||||||||||||||||
 マップデータ マップデータ | primary map | |||||||||||||||||||||
 試料 試料 |
| |||||||||||||||||||||
 キーワード キーワード | Protease / HYDROLASE | |||||||||||||||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報endopeptidase Clp / endopeptidase Clp complex / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / ATPase binding / serine-type endopeptidase activity / identical protein binding / cytoplasm 類似検索 - 分子機能 | |||||||||||||||||||||
| 生物種 |  Staphylococcus aureus (strain Newman) (黄色ブドウ球菌) / Staphylococcus aureus subsp. aureus str. Newman (黄色ブドウ球菌) Staphylococcus aureus (strain Newman) (黄色ブドウ球菌) / Staphylococcus aureus subsp. aureus str. Newman (黄色ブドウ球菌) | |||||||||||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.7 Å | |||||||||||||||||||||
 データ登録者 データ登録者 | Ripstein ZA / Vahidi S / Kay LE / Rubinstein JL | |||||||||||||||||||||
| 資金援助 |  カナダ, 6件 カナダ, 6件
| |||||||||||||||||||||
 引用 引用 | ジャーナル: Proc Natl Acad Sci U S A / 年: 2018 タイトル: Reversible inhibition of the ClpP protease via an N-terminal conformational switch. 著者: Siavash Vahidi / Zev A Ripstein / Massimiliano Bonomi / Tairan Yuwen / Mark F Mabanglo / Jordan B Juravsky / Kamran Rizzolo / Algirdas Velyvis / Walid A Houry / Michele Vendruscolo / John L ...著者: Siavash Vahidi / Zev A Ripstein / Massimiliano Bonomi / Tairan Yuwen / Mark F Mabanglo / Jordan B Juravsky / Kamran Rizzolo / Algirdas Velyvis / Walid A Houry / Michele Vendruscolo / John L Rubinstein / Lewis E Kay /  要旨: Protein homeostasis is critically important for cell viability. Key to this process is the refolding of misfolded or aggregated proteins by molecular chaperones or, alternatively, their degradation ...Protein homeostasis is critically important for cell viability. Key to this process is the refolding of misfolded or aggregated proteins by molecular chaperones or, alternatively, their degradation by proteases. In most prokaryotes and in chloroplasts and mitochondria, protein degradation is performed by the caseinolytic protease ClpP, a tetradecamer barrel-like proteolytic complex. Dysregulating ClpP function has shown promise in fighting antibiotic resistance and as a potential therapy for acute myeloid leukemia. Here we use methyl-transverse relaxation-optimized spectroscopy (TROSY)-based NMR, cryo-EM, biochemical assays, and molecular dynamics simulations to characterize the structural dynamics of ClpP from (SaClpP) in wild-type and mutant forms in an effort to discover conformational hotspots that regulate its function. Wild-type SaClpP was found exclusively in the active extended form, with the N-terminal domains of its component protomers in predominantly β-hairpin conformations that are less well-defined than other regions of the protein. A hydrophobic site was identified that, upon mutation, leads to unfolding of the N-terminal domains, loss of SaClpP activity, and formation of a previously unobserved split-ring conformation with a pair of 20-Å-wide pores in the side of the complex. The extended form of the structure and partial activity can be restored via binding of ADEP small-molecule activators. The observed structural plasticity of the N-terminal gates is shown to be a conserved feature through studies of and ClpP, suggesting a potential avenue for the development of molecules to allosterically modulate the function of ClpP. | |||||||||||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
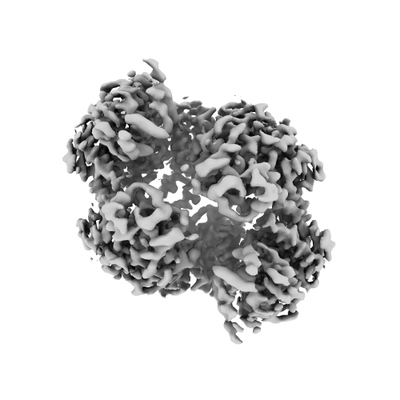
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_7952.map.gz | 3 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-7952-v30.xmlemd-7952.xml | 16.6 KB 16.6 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 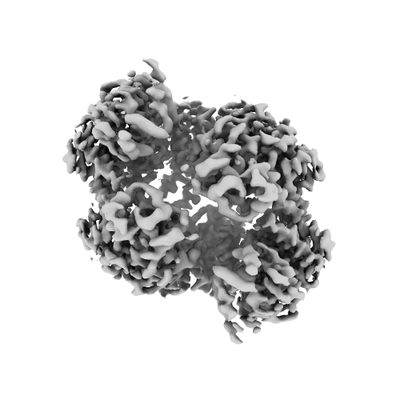 emd_7952.png emd_7952.png | 48.3 KB | ||
| Filedesc metadata | emd-7952.cif.gz | 6 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7952 http://ftp.pdbj.org/pub/emdb/structures/EMD-7952ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7952 | HTTPS FTP |
-関連構造データ








-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_7952.map.gz / 形式: CCP4 / 大きさ: 27 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | primary map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.06 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
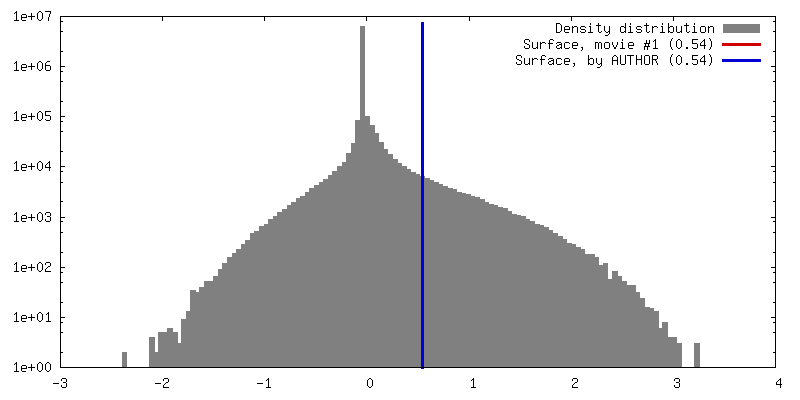
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Caseinolytic protease from Staphylococcus aureus (V7A)
| 全体 | 名称: Caseinolytic protease from Staphylococcus aureus (V7A) |
|---|---|
| 要素 |
|
-超分子 #1: Caseinolytic protease from Staphylococcus aureus (V7A)
| 超分子 | 名称: Caseinolytic protease from Staphylococcus aureus (V7A) タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: all |
|---|---|
| 由来(天然) | 生物種: Staphylococcus aureus (strain Newman) (黄色ブドウ球菌) 株: Newman |
| 分子量 | 理論値: 301 KDa |
-分子 #1: ATP-dependent Clp protease proteolytic subunit
| 分子 | 名称: ATP-dependent Clp protease proteolytic subunit / タイプ: protein_or_peptide / ID: 1 / コピー数: 14 / 光学異性体: LEVO / EC番号: endopeptidase Clp |
|---|---|
| 由来(天然) | 生物種: Staphylococcus aureus subsp. aureus str. Newman (黄色ブドウ球菌) 株: Newman |
| 分子量 | 理論値: 21.508479 KDa |
| 組換発現 | 生物種: |
| 配列 | 文字列: MNLIPTAIET TNRGERAYDI YSRLLKDRII MLGSQIDDNV ANSIVSQLLF LQAQDSEKDI YLYINSPGGS VTAGFAIYDT IQHIKPDVQ TICIGMAASM GSFLLAAGAK GKRFALPNAE VMIHQPLGGA QGQATEIEIA ANHILKTREK LNRILSERTG Q SIEKIQKD ...文字列: MNLIPTAIET TNRGERAYDI YSRLLKDRII MLGSQIDDNV ANSIVSQLLF LQAQDSEKDI YLYINSPGGS VTAGFAIYDT IQHIKPDVQ TICIGMAASM GSFLLAAGAK GKRFALPNAE VMIHQPLGGA QGQATEIEIA ANHILKTREK LNRILSERTG Q SIEKIQKD TDRDNFLTAE EAKEYGLIDE VMVPETK UniProtKB: ATP-dependent Clp protease proteolytic subunit |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 30 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 7 構成要素:
| ||||||||||||||||||
| グリッド | 支持フィルム - トポロジー: HOLEY / 支持フィルム - Film thickness: 35 / 詳細: unspecified | ||||||||||||||||||
| 凍結 | 凍結剤: ETHANE-PROPANE / チャンバー内湿度: 100 % / チャンバー内温度: 277 K / 装置: FEI VITROBOT MARK III / 詳細: Modified Vitrobot. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: FEI FALCON III (4k x 4k) 検出モード: COUNTING / デジタル化 - サイズ - 横: 4096 pixel / デジタル化 - サイズ - 縦: 4096 pixel / デジタル化 - 画像ごとのフレーム数: 1-30 / 撮影したグリッド数: 2 / 実像数: 1837 / 平均露光時間: 60.0 sec. / 平均電子線量: 43.0 e/Å2 / 詳細: movies were collected with 44 fractions |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 100.0 µm / 最大 デフォーカス(補正後): 3.2 µm / 最小 デフォーカス(補正後): 1.0 µm / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 倍率(公称値): 75000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
+画像解析
-原子モデル構築 1
| 初期モデル |
| ||||||
|---|---|---|---|---|---|---|---|
| 精密化 | 空間: REAL / プロトコル: OTHER | ||||||
| 得られたモデル |  PDB-6dkf: |


