Journal: Elife / Year: 2016 Title: Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Authors: Corey M Dambacher / Evan J Worden / Mark A Herzik / Andreas Martin / Gabriel C Lander / Abstract: The 26S proteasome is responsible for the selective, ATP-dependent degradation of polyubiquitinated cellular proteins. Removal of ubiquitin chains from targeted substrates at the proteasome is a ...The 26S proteasome is responsible for the selective, ATP-dependent degradation of polyubiquitinated cellular proteins. Removal of ubiquitin chains from targeted substrates at the proteasome is a prerequisite for substrate processing and is accomplished by Rpn11, a deubiquitinase within the 'lid' sub-complex. Prior to the lid's incorporation into the proteasome, Rpn11 deubiquitinase activity is inhibited to prevent unwarranted deubiquitination of polyubiquitinated proteins. Here we present the atomic model of the isolated lid sub-complex, as determined by cryo-electron microscopy at 3.5 Å resolution, revealing how Rpn11 is inhibited through its interaction with a neighboring lid subunit, Rpn5. Through mutagenesis of specific residues, we describe the network of interactions that are required to stabilize this inhibited state. These results provide significant insight into the intricate mechanisms of proteasome assembly, outlining the substantial conformational rearrangements that occur during incorporation of the lid into the 26S holoenzyme, which ultimately activates the deubiquitinase for substrate degradation.
History
Deposition
Oct 11, 2015
-
Header (metadata) release
Nov 11, 2015
-
Map release
Jan 20, 2016
-
Update
May 11, 2016
-
Current status
May 11, 2016
Processing site: RCSB / Status: Released
-
Structure visualization
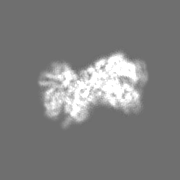
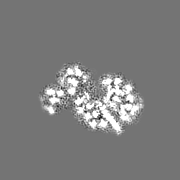
Movie
Surface view with section colored by density value
Entire : Recombinant yeast 26S proteasome lid complex
Entire
Name: Recombinant yeast 26S proteasome lid complex
Components
Sample: Recombinant yeast 26S proteasome lid complex
Protein or peptide: 26S proteasome lid sub-complex
-
Supramolecule #1000: Recombinant yeast 26S proteasome lid complex
Supramolecule
Name: Recombinant yeast 26S proteasome lid complex / type: sample / ID: 1000 / Details: The sample was monodisperse. / Oligomeric state: 9 subunits / Number unique components: 1
Molecular weight
Experimental: 370 KDa / Theoretical: 370 KDa / Method: SDS protein gels and size exclusion chromatography
-
Macromolecule #1: 26S proteasome lid sub-complex
Macromolecule
Name: 26S proteasome lid sub-complex / type: protein_or_peptide / ID: 1 / Name.synonym: lid Details: Lid complex was recombinantly expressed in E. coli and purified by size exclusion chromatography. Number of copies: 1 / Oligomeric state: Heterononamer / Recombinant expression: Yes
GO: proteasome regulatory particle, lid subcomplex / InterPro: Proteasome/cyclosome repeat
-
Experimental details
-
Structure determination
Method
cryo EM
Processing
single particle reconstruction
Aggregation state
particle
-
Sample preparation
Concentration
2.5 mg/mL
Buffer
pH: 7.5 / Details: 50 mM HEPES, 100 mM NaCl, 100 mM KCl, 1 mM TCEP
Grid
Details: Sample was applied directly to plasma-cleaned holey carbon C-flat grids (400 mesh, 1.2 micrometer holes).
Vitrification
Cryogen name: ETHANE / Chamber humidity: 88 % / Chamber temperature: 85 K / Instrument: HOMEMADE PLUNGER / Details: Manual plunging was performed in a cold room. Method: 4 microliters of sample was applied to the grid, blotted for 2 seconds, and plunged into liquid ethane.
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Temperature
Min: 85 K / Max: 90 K / Average: 87.5 K
Alignment procedure
Legacy - Astigmatism: Objective lens astigmatism was corrected at a nominal magnification of 22,500.
Details
Micrographs were collected in super-resolution mode with a total frame count of 38 and total exposure time of 7.6 seconds.
Date
Feb 10, 2015
Image recording
Category: CCD / Film or detector model: GATAN K2 (4k x 4k) / Number real images: 3432 / Average electron dose: 43.8 e/Å2 Details: Micrographs were collected as movies using super-resolution mode with the Gatan K2 Summit direct electron detector
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Image pre-processing was performed using Appion. 3D classification and reconstruction was performed with RELION.
CTF correction
Details: whole micrograph
Final reconstruction
Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 3.5 Å / Resolution method: OTHER / Software - Name: Appion, CTFFIND3, FindEM, RELION Details: 3D classification was performed to identify the best 109,396 particles from an initial data set of 254,112. Number images used: 109396
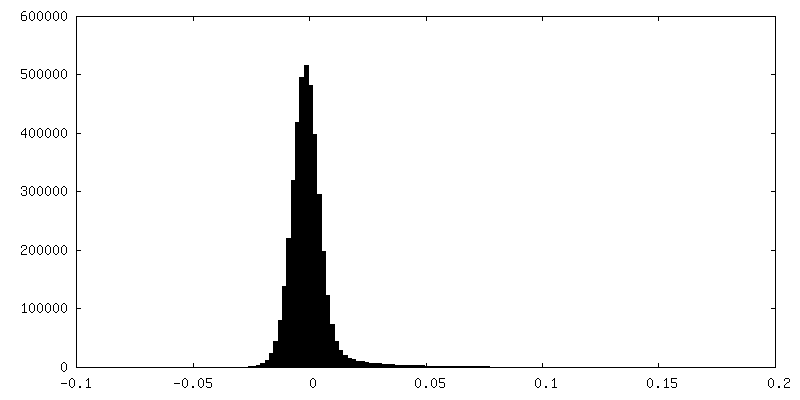
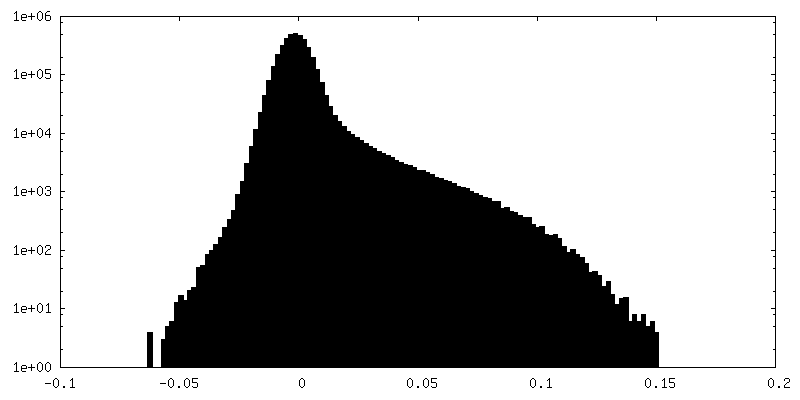
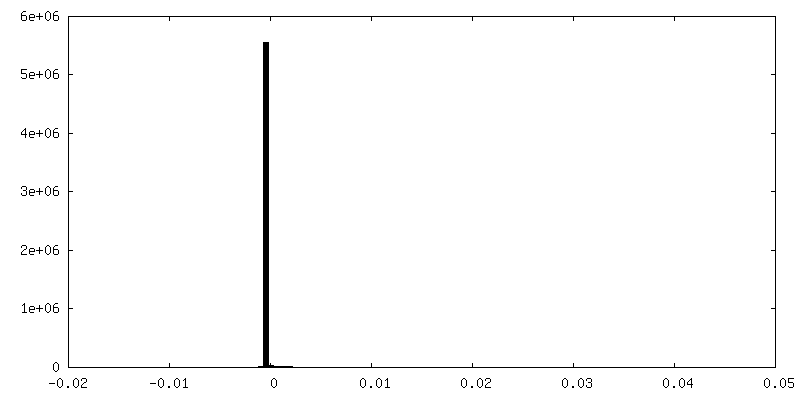
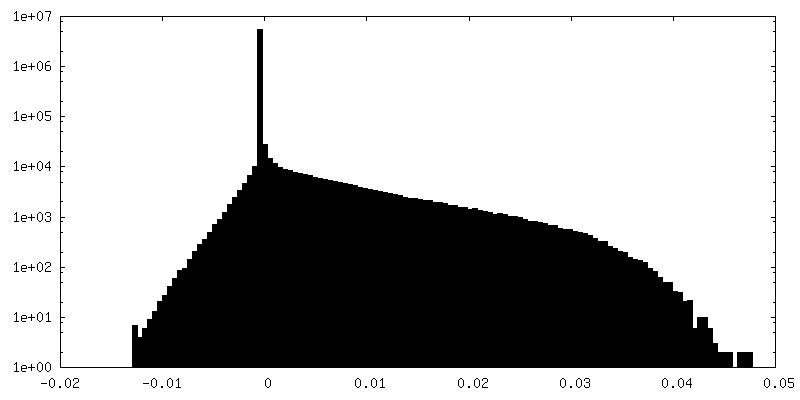
FSC plot (resolution estimation)
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
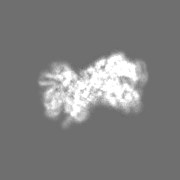
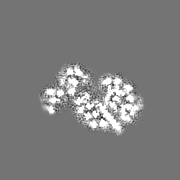
Downloads & links 400_6479.gif
400_6479.gif 80_6479.gif
80_6479.gif http://ftp.pdbj.org/pub/emdb/structures/EMD-6479
http://ftp.pdbj.org/pub/emdb/structures/EMD-6479
























 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)































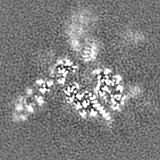
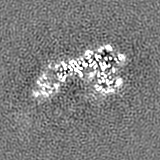
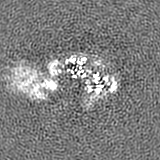








 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN

