 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3571 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
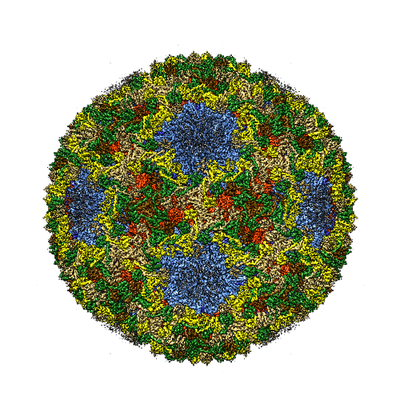
| Title | dsRNA bacteriophage phi6 nucleocapsid | |||||||||
 Map data Map data | dsRNA bacteriophage phi6 nucleocapsid | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | icosahedral virus capsid shell / virus | |||||||||
| Function / homology |  Function and homology information Function and homology informationT=13 icosahedral viral capsid / viral procapsid / T=2 icosahedral viral capsid / viral genome packaging / viral inner capsid / viral outer capsid / viral capsid / ribonucleoside triphosphate phosphatase activity / nucleoside-triphosphate phosphatase / viral nucleocapsid ...T=13 icosahedral viral capsid / viral procapsid / T=2 icosahedral viral capsid / viral genome packaging / viral inner capsid / viral outer capsid / viral capsid / ribonucleoside triphosphate phosphatase activity / nucleoside-triphosphate phosphatase / viral nucleocapsid / DNA-templated transcription / RNA binding / ATP binding / identical protein binding Similarity search - Function | |||||||||
| Biological species |  Pseudomonas phage phi6 (virus) Pseudomonas phage phi6 (virus) | |||||||||
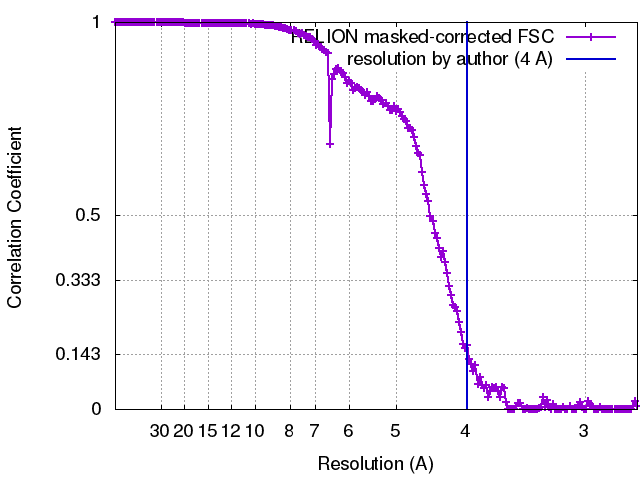
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | |||||||||
 Authors Authors | Sun Z / El Omari K | |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2017 Title: Double-stranded RNA virus outer shell assembly by bona fide domain-swapping. Authors: Zhaoyang Sun / Kamel El Omari / Xiaoyu Sun / Serban L Ilca / Abhay Kotecha / David I Stuart / Minna M Poranen / Juha T Huiskonen /   Abstract: Correct outer protein shell assembly is a prerequisite for virion infectivity in many multi-shelled dsRNA viruses. In the prototypic dsRNA bacteriophage φ6, the assembly reaction is promoted by ...Correct outer protein shell assembly is a prerequisite for virion infectivity in many multi-shelled dsRNA viruses. In the prototypic dsRNA bacteriophage φ6, the assembly reaction is promoted by calcium ions but its biomechanics remain poorly understood. Here, we describe the near-atomic resolution structure of the φ6 double-shelled particle. The outer T=13 shell protein P8 consists of two alpha-helical domains joined by a linker, which allows the trimer to adopt either a closed or an open conformation. The trimers in an open conformation swap domains with each other. Our observations allow us to propose a mechanistic model for calcium concentration regulated outer shell assembly. Furthermore, the structure provides a prime exemplar of bona fide domain-swapping. This leads us to extend the theory of domain-swapping from the level of monomeric subunits and multimers to closed spherical shells, and to hypothesize a mechanism by which closed protein shells may arise in evolution. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3571.map.gz | 464.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3571-v30.xmlemd-3571.xml | 18.2 KB 18.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3571_fsc.xml | 17.8 KB | Display | FSC data file |
| Images |  emd_3571.png emd_3571.png | 216.1 KB | ||
| Filedesc metadata | emd-3571.cif.gz | 7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3571ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3571 http://ftp.pdbj.org/pub/emdb/structures/EMD-3571ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3571 | HTTPS FTP |
-Related structure data
| Related structure data |  5muuMC  3572C  3573C  5muvC  5muwC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_3571.map.gz / Format: CCP4 / Size: 512 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | dsRNA bacteriophage phi6 nucleocapsid | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Pseudomonas phage phi6
| Entire | Name: Pseudomonas phage phi6 (virus) |
|---|---|
| Components |
|
-Supramolecule #1: Pseudomonas phage phi6
| Supramolecule | Name: Pseudomonas phage phi6 / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all Details: The viral envelope was removed by Triton X-114 extraction NCBI-ID: 10879 / Sci species name: Pseudomonas phage phi6 / Virus type: VIRION / Virus isolate: SPECIES / Virus enveloped: Yes / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Pseudomonas syringae (bacteria) / Strain: pv.phaseolicola HB10Y Pseudomonas syringae (bacteria) / Strain: pv.phaseolicola HB10Y |
| Virus shell | Shell ID: 1 / Name: Outer shell / Diameter: 565.0 Å / T number (triangulation number): 13 |
| Virus shell | Shell ID: 2 / Name: Inner shell / Diameter: 500.0 Å / T number (triangulation number): 1 |
-Macromolecule #1: Major inner protein P1
| Macromolecule | Name: Major inner protein P1 / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Pseudomonas phage phi6 (virus) |
| Molecular weight | Theoretical: 85.080711 KDa |
| Sequence | String: MFNLKVKDLN GSARGLTQAF AIGELKNQLS VGALQLPLQF TRTFSASMTS ELLWEVGKGN IDPVMYARLF FQYAQAGGAL SVDELVNQF TEYHQSTACN PEIWRKLTAY ITGSSNRAIK ADAVGKVPPT AILEQLRTLA PSEHELFHHI TTDFVCHVLS P LGFILPDA ...String: MFNLKVKDLN GSARGLTQAF AIGELKNQLS VGALQLPLQF TRTFSASMTS ELLWEVGKGN IDPVMYARLF FQYAQAGGAL SVDELVNQF TEYHQSTACN PEIWRKLTAY ITGSSNRAIK ADAVGKVPPT AILEQLRTLA PSEHELFHHI TTDFVCHVLS P LGFILPDA AYVYRVGRTA TYPNFYALVD CVRASDLRRM LTALSSVDSK MLQATFKAKG ALAPALISQH LANAATTAFE RS RGNFDAN AVVSSVLTIL GRLWSPSTPK ELDPSARLRN TNGIDQLRSN LALFIAYQDM VKQRGRAEVI FSDEELSSTI IPW FIEAMS EVSPFKLRPI NETTSYIGQT SAIDHMGQPS HVVVYEDWQF AKEITAFTPV KLANNSNQRF LDVEPGISDR MSAT LAPIG NTFAVSAFVK NRTAVYEAVS QRGTVNSNGA EMTLGFPSVV ERDYALDRDP MVAIAALRTG IVDESLEARA SNDLK RSMF NYYAAVMHYA VAHNPEVVVS EHQGVAAEQG SLYLVWNVRT ELRIPVGYNA IEGGSIRTPE PLEAIAYNKP IQPSEV LQA KVLDLANHTT SIHIWPWHEA STEFAYEDAY SVTIRNKRYT AEVKEFELLG LGQRRERVRI LKPTVAHAII QMWYSWF VE DDRTLAAARR TSRDDAEKLA IDGRRMQNAV TLLRKIEMIG TTGIGASAVH LAQSRIVDQM AGRGLIDDSS DLHVGINR H RIRIWAGLAV LQMMGLLSRS EAEALTKVLG DSNALGMVVA TTDIDPSL UniProtKB: Major inner protein P1 |
-Macromolecule #2: Packaging enzyme P4
| Macromolecule | Name: Packaging enzyme P4 / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO / EC number: nucleoside-triphosphate phosphatase |
|---|---|
| Source (natural) | Organism: Pseudomonas phage phi6 (virus) |
| Molecular weight | Theoretical: 35.198426 KDa |
| Sequence | String: MPIVVTQAHI DRVGIAADLL DASPVSLQVL GRPTAINTVV IKTYIAAVME LASKQGGSLA GVDIRPSVLL KDTAIFTKPK AKSADVESD VDVLDTGIYS VPGLARKPVT HRWPSEGIYS GVTALMGATG SGKSITLNEK LRPDVLIRWG EVAEAYDELD T AVHISTLD ...String: MPIVVTQAHI DRVGIAADLL DASPVSLQVL GRPTAINTVV IKTYIAAVME LASKQGGSLA GVDIRPSVLL KDTAIFTKPK AKSADVESD VDVLDTGIYS VPGLARKPVT HRWPSEGIYS GVTALMGATG SGKSITLNEK LRPDVLIRWG EVAEAYDELD T AVHISTLD EMLIVCIGLG ALGFNVAVDS VRPLLFRLKG AASAGGIVAV FYSLLTDISN LFTQYDCSVV MVVNPMVDAE KI EYVFGQV MASTVGAILC ADGNVSRTMF RTNKGRIFNG AAPLAADTHM PSMDRPTSMK ALDHTSIASV APLERGSVDT DDR NSAPRR GANFSL UniProtKB: Packaging enzyme P4 |
-Macromolecule #3: Major outer capsid protein
| Macromolecule | Name: Major outer capsid protein / type: protein_or_peptide / ID: 3 / Number of copies: 10 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Pseudomonas phage phi6 (virus) |
| Molecular weight | Theoretical: 16.018418 KDa |
| Sequence | String: MLLPVVARAA VPAIESAIAA TPGLVSRIAA AIGSKVSPSA ILAAVKSNPV VAGLTLAQIG STGYDAYQQL LENHPEVAEM LKDLSFKAD EIQPDFIGNL GQYREELELV EDAARFVGGM SNLIRLRQAL ELDIKYYGLK MQLNDMGYRS UniProtKB: Major outer capsid protein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3 mg/mL |
|---|---|
| Buffer | pH: 7.2 |
| Grid | Model: C-flat / Material: COPPER / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 15 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK III |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Temperature | Min: 80.0 K / Max: 120.0 K |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Lower energy threshold: 0 eV / Energy filter - Upper energy threshold: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-22 / Number real images: 900 / Average exposure time: 0.2 sec. / Average electron dose: 0.73 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: 3.0 µm / Calibrated magnification: 37037 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 0.3 µm / Nominal magnification: 160000 |
| Sample stage | Specimen holder model: OTHER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 1-760 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | The structure of P1 was fitted in the map using COOT as a rigid body in two different positions corresponding to subunits P1A and P1B. P1A and P1B main-chains and side-chains were adjusted using manual and real space fitting in COOT. Structure was refined in Phenix.real_space_refine applying secondary structure, rotamer, and Ramachandran plot restraints. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-5muu: |

-Atomic model buiding 2
| Details | The structure of P8 (L3 to Y147) was built manually in COOT and side-chains were adjusted using manual and real space fitting in COOT. Structure was refined in Phenix.real_space_refine applying secondary structure, rotamer, and Ramachandran plot restraints. |
|---|---|
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
| Output model | PDB-5muu: |
-Atomic model buiding 3
| Details | The structure of P4 C-terminus (R292 to L332) was built manually in COOT and side-chains were adjusted using manual and real space fitting in COOT. Structure was refined in Phenix.real_space_refine applying secondary structure, rotamer, and Ramachandran plot restraints. |
|---|---|
| Refinement | Protocol: AB INITIO MODEL |
| Output model | PDB-5muu: |
