 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
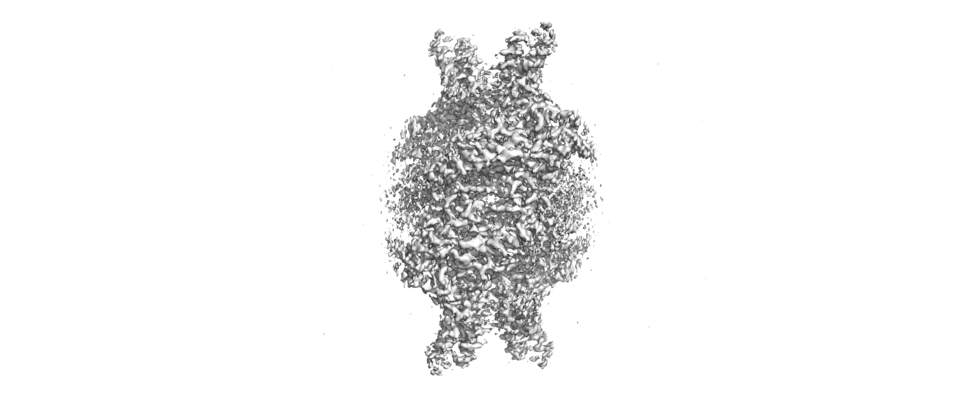
| Entry | Database: EMDB / ID: EMD-31053 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of Isocitrate lyase-1 from Candida albicans | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Isocitrate lyase / glyoxylate cycle / ubiquitination / LYASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationisocitrate lyase activity / methylisocitrate lyase activity / glyoxylate cycle / tricarboxylic acid cycle / peroxisome / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Candida albicans (yeast) Candida albicans (yeast) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.63 Å | |||||||||
 Authors Authors | Hiragi K / Nishio K | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2021 Title: Structural insights into the targeting specificity of ubiquitin ligase for S. cerevisiae isocitrate lyase but not C. albicans isocitrate lyase. Authors: Keito Hiragi / Kazuya Nishio / Shu Moriyama / Tasuku Hamaguchi / Akira Mizoguchi / Koji Yonekura / Kazutoshi Tani / Tsunehiro Mizushima /  Abstract: In Saccharomyces cerevisiae, the glyoxylate cycle is controlled through the posttranslational regulation of its component enzymes, such as isocitrate lyase (ICL), which catalyzes the first unique ...In Saccharomyces cerevisiae, the glyoxylate cycle is controlled through the posttranslational regulation of its component enzymes, such as isocitrate lyase (ICL), which catalyzes the first unique step of the cycle. The ICL of S.cerevisiae (ScIcl1) is tagged for proteasomal degradation through ubiquitination by a multisubunit ubiquitin ligase (the glucose-induced degradation-deficient (GID) complex), whereas that of the pathogenic yeast Candida albicans (CaIcl1) escapes this process. However, the reason for the ubiquitin targeting specificity of the GID complex for ScIcl1 and not for CaIcl1 is unclear. To gain some insight into this, in this study, the crystal structures of apo ScIcl1 and CaIcl1 in complex with formate and the cryogenic electron microscopy structure of apo CaIcl1 were determined at a resolution of 2.3, 2.7, and 2.6 Å, respectively. A comparison of the various structures suggests that the orientation of N-terminal helix α1 in S.cerevisiae is likely key to repositioning of ubiquitination sites and contributes to the distinction found in C. albicans ubiquitin evasion mechanism. This finding gives us a better understanding of the molecular mechanism of ubiquitin-dependent ScIcl1 degradation and could serve as a theoretical basis for the research and development of anti-C. albicans drugs based on the concept of CaIcl1 ubiquitination. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_31053.map.gz | 116.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-31053-v30.xmlemd-31053.xml | 11.3 KB 11.3 KB | Display Display | EMDB header |
| Images |  emd_31053.png emd_31053.png | 86.3 KB | ||
| Filedesc metadata | emd-31053.cif.gz | 5.5 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-31053ftp://ftp.pdbj.org/pub/emdb/structures/EMD-31053 http://ftp.pdbj.org/pub/emdb/structures/EMD-31053ftp://ftp.pdbj.org/pub/emdb/structures/EMD-31053 | HTTPS FTP |
-Related structure data
| Related structure data |  7ebfMC  7ebcC  7ebeC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_31053.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.823 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Isocitrate lyase
| Entire | Name: Isocitrate lyase |
|---|---|
| Components |
|
-Supramolecule #1: Isocitrate lyase
| Supramolecule | Name: Isocitrate lyase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Candida albicans (yeast) |
-Macromolecule #1: Isocitrate lyase
| Macromolecule | Name: Isocitrate lyase / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO / EC number: isocitrate lyase |
|---|---|
| Source (natural) | Organism: Candida albicans (yeast) |
| Molecular weight | Theoretical: 62.336523 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: HHHHHHMPYT PIDIQKEEAD FQKEVAEIKK WWSEPRWRKT KRIYSAEDIA KKRGTLKINH PSSQQADKLF KLLEKHDADK TVSFTFGAL DPIHVAQMAK YLDSIYVSGW QCSSTASTSN EPSPDLADYP MDTVPNKVEH LWFAQLFHDR KQREERLTLS K EERAKTPY ...String: HHHHHHMPYT PIDIQKEEAD FQKEVAEIKK WWSEPRWRKT KRIYSAEDIA KKRGTLKINH PSSQQADKLF KLLEKHDADK TVSFTFGAL DPIHVAQMAK YLDSIYVSGW QCSSTASTSN EPSPDLADYP MDTVPNKVEH LWFAQLFHDR KQREERLTLS K EERAKTPY IDFLRPIIAD ADTGHGGITA IIKLTKMFIE RGAAGIHIED QAPGTKKCGH MAGKVLVPVQ EHINRLVAIR AS ADIFGSN LLAVARTDSE AATLITSTID HRDHYFIIGA TNPEAGDLAA LMAEAESKGI YGNELAAIES EWTKKAGLKL FHE AVIDEI KNGNYSNKDA LIKKFTDKVN PLSHTSHKEA KKLAKELTGK DIYFNWDVAR AREGYYRYQG GTQCAVMRGR AFAP YADLI WMESALPDYA QAKEFADGVK AAVPDQWLAY NLSPSFNWNK AMPADEQETY IKRLGKLGYV WQFITLAGLH TTALA VDDF SNQYSQIGMK AYGQTVQQPE IEKGVEVVKH QKWSGATYID GLLKMVSGGV TSTAAMGQGV TEDQFKESKA KA UniProtKB: Isocitrate lyase |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 278 K |
- Electron microscopy
Electron microscopy
| Microscope | JEOL CRYO ARM 300 |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average exposure time: 2.0 sec. / Average electron dose: 42.7 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
