Journal: Cell Rep / Year: 2015 Title: Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Authors: Ola B Nilsson / Rickard Hedman / Jacopo Marino / Stephan Wickles / Lukas Bischoff / Magnus Johansson / Annika Müller-Lucks / Fabio Trovato / Joseph D Puglisi / Edward P O'Brien / Roland ...Authors: Ola B Nilsson / Rickard Hedman / Jacopo Marino / Stephan Wickles / Lukas Bischoff / Magnus Johansson / Annika Müller-Lucks / Fabio Trovato / Joseph D Puglisi / Edward P O'Brien / Roland Beckmann / Gunnar von Heijne / Abstract: At what point during translation do proteins fold? It is well established that proteins can fold cotranslationally outside the ribosome exit tunnel, whereas studies of folding inside the exit tunnel ...At what point during translation do proteins fold? It is well established that proteins can fold cotranslationally outside the ribosome exit tunnel, whereas studies of folding inside the exit tunnel have so far detected only the formation of helical secondary structure and collapsed or partially structured folding intermediates. Here, using a combination of cotranslational nascent chain force measurements, inter-subunit fluorescence resonance energy transfer studies on single translating ribosomes, molecular dynamics simulations, and cryoelectron microscopy, we show that a small zinc-finger domain protein can fold deep inside the vestibule of the ribosome exit tunnel. Thus, for small protein domains, the ribosome itself can provide the kind of sheltered folding environment that chaperones provide for larger proteins.
History
Deposition
Jul 8, 2015
-
Header (metadata) release
Aug 12, 2015
-
Map release
Sep 9, 2015
-
Update
Sep 23, 2015
-
Current status
Sep 23, 2015
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information

 Authors
Authors Citation
Citation

 Structure visualization
Structure visualization
 Downloads & links
Downloads & links http://ftp.pdbj.org/pub/emdb/structures/EMD-3079
http://ftp.pdbj.org/pub/emdb/structures/EMD-3079

























 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

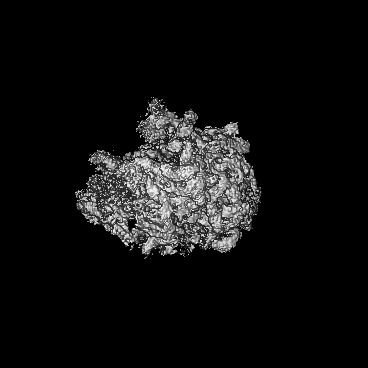














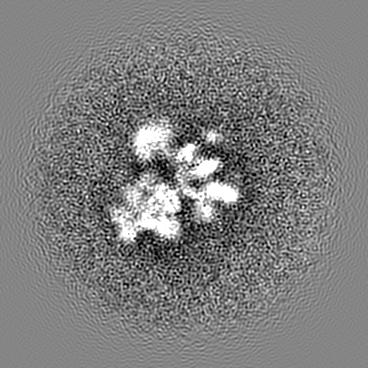




 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN

