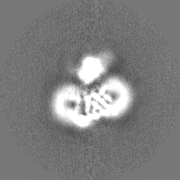
Journal: Protein Sci / Year: 2020 Title: Consensus mutagenesis approach improves the thermal stability of system x transporter, xCT, and enables cryo-EM analyses. Authors: Kazumasa Oda / Yongchan Lee / Pattama Wiriyasermkul / Yoko Tanaka / Mizuki Takemoto / Keitaro Yamashita / Shushi Nagamori / Tomohiro Nishizawa / Osamu Nureki / Abstract: System x is an amino acid antiporter that imports L-cystine into cells and exports intracellular L-glutamate, at a 1:1 ratio. As L-cystine is an essential precursor for glutathione synthesis, system ...System x is an amino acid antiporter that imports L-cystine into cells and exports intracellular L-glutamate, at a 1:1 ratio. As L-cystine is an essential precursor for glutathione synthesis, system x supports tumor cell growth through glutathione-based oxidative stress resistance and is considered as a potential therapeutic target for cancer treatment. System x consists of two subunits, the light chain subunit SLC7A11 (xCT) and the heavy chain subunit SLC3A2 (also known as CD98hc or 4F2hc), which are linked by a conserved disulfide bridge. Although the recent structures of another SLC7 member, L-type amino acid transporter 1 (LAT1) in complex with CD98hc, have provided the structural basis toward understanding the amino acid transport mechanism, the detailed molecular mechanism of xCT remains unknown. To revealthe molecular mechanism, we performed single-particle analyses of the xCT-CD98hc complex. As wild-type xCT-CD98hc displayed poor stability and could not be purified to homogeneity, we applied a consensus mutagenesis approach to xCT. The consensus mutated construct exhibited increased stability as compared to the wild-type, and enabled the cryoelectron microscopy (cryo-EM) map to be obtained at 6.2 Å resolution by single-particle analysis. The cryo-EM map revealed sufficient electron density to assign secondary structures. In the xCT structure, the hash and arm domains are well resolved, whereas the bundle domain shows some flexibility. CD98hc is positioned next to the xCT transmembrane domain. This study provides the structural basis of xCT, and our consensus-based strategy could represent a good choice toward solving unstable protein structures.
History
Deposition
Jun 17, 2020
-
Header (metadata) release
Dec 9, 2020
-
Map release
Dec 9, 2020
-
Update
Nov 20, 2024
-
Current status
Nov 20, 2024
Processing site: PDBj / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 49.76 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Electron optics
Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
+
Image processing
Startup model
Type of model: OTHER
Final reconstruction
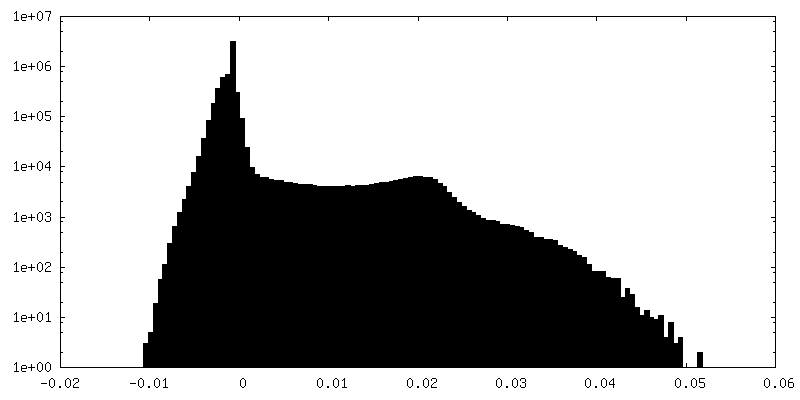
Resolution.type: BY AUTHOR / Resolution: 6.2 Å / Resolution method: FSC 0.143 CUT-OFF Details: This pdb entry contains an issue about peptide linkage (bond outlier). Residues GLU A 213 and LEU A 214 that are next to each other are not properly linked: distance between C and N is 3.35. ...Details: This pdb entry contains an issue about peptide linkage (bond outlier). Residues GLU A 213 and LEU A 214 that are next to each other are not properly linked: distance between C and N is 3.35. This was introduced upon the fitting of the homology model into the low resolution cryo-EM map by using Rosetta. The model has not undergone any further structure refinement. Number images used: 86395
Initial angle assignment
Type: MAXIMUM LIKELIHOOD
Final angle assignment
Type: MAXIMUM LIKELIHOOD
FSC plot (resolution estimation)
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) Authors
Authors Citation
Citation

 Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_30341.png
emd_30341.png http://ftp.pdbj.org/pub/emdb/structures/EMD-30341
http://ftp.pdbj.org/pub/emdb/structures/EMD-30341












 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)











































 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN

