 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-23271 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
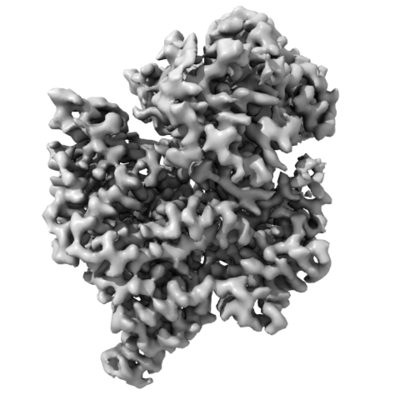
| Title | Helitron transposase bound to LTS | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Helitron / transposase / evolution / gene editing / gene capture / rolling circle / replication / recombination / nuclease / helicase / relaxase | |||||||||
| Biological species | synthetic construct (others) /  Myotis lucifugus (little brown bat) Myotis lucifugus (little brown bat) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.66 Å | |||||||||
 Authors Authors | Kosek D / Dyda F | |||||||||
| Funding support | 1 items
| |||||||||
 Citation Citation | Journal: Mol Cell / Year: 2021 Title: The large bat Helitron DNA transposase forms a compact monomeric assembly that buries and protects its covalently bound 5'-transposon end. Authors: Dalibor Kosek / Ivana Grabundzija / Haotian Lei / Ilija Bilic / Huaibin Wang / Yukun Jin / Graham F Peaslee / Alison B Hickman / Fred Dyda /   Abstract: Helitrons are widespread eukaryotic DNA transposons that have significantly contributed to genome variability and evolution, in part because of their distinctive, replicative rolling-circle ...Helitrons are widespread eukaryotic DNA transposons that have significantly contributed to genome variability and evolution, in part because of their distinctive, replicative rolling-circle mechanism, which often mobilizes adjacent genes. Although most eukaryotic transposases form oligomers and use RNase H-like domains to break and rejoin double-stranded DNA (dsDNA), Helitron transposases contain a single-stranded DNA (ssDNA)-specific HUH endonuclease domain. Here, we report the cryo-electron microscopy structure of a Helitron transposase bound to the 5'-transposon end, providing insight into its multidomain architecture and function. The monomeric transposase forms a tightly packed assembly that buries the covalently attached cleaved end, protecting it until the second end becomes available. The structure reveals unexpected architectural similarity to TraI, a bacterial relaxase that also catalyzes ssDNA movement. The HUH active site suggests how two juxtaposed tyrosines, a feature of many replication initiators that use HUH nucleases, couple the conformational shift of an α-helix to control strand cleavage and ligation reactions. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_23271.map.gz | 28.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-23271-v30.xmlemd-23271.xml | 13.7 KB 13.7 KB | Display Display | EMDB header |
| Images |  emd_23271.png emd_23271.png | 103.5 KB | ||
| Filedesc metadata | emd-23271.cif.gz | 6.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23271ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23271 http://ftp.pdbj.org/pub/emdb/structures/EMD-23271ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23271 | HTTPS FTP |
-Related structure data
| Related structure data |  7lccMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_23271.map.gz / Format: CCP4 / Size: 40.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
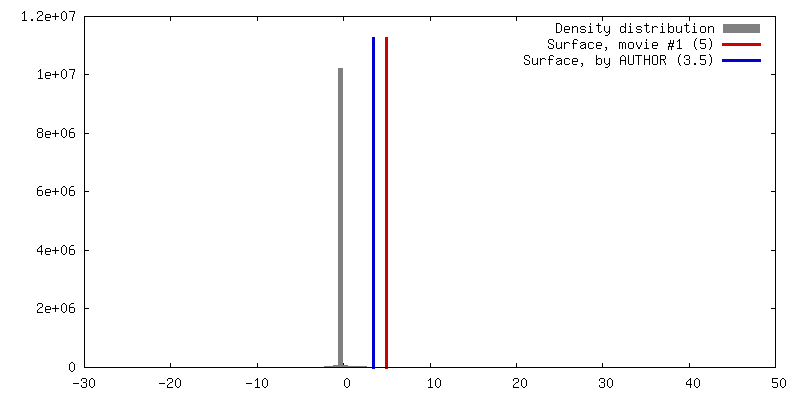
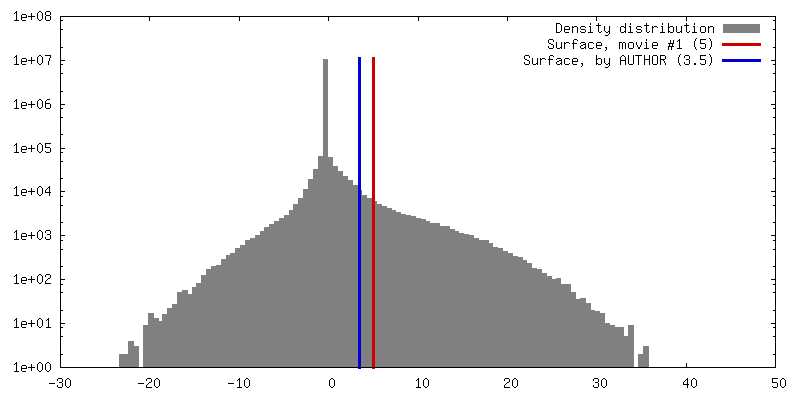
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)


















-Supplemental data
- Sample components
Sample components
-Entire : Helraiser complex with LTS
| Entire | Name: Helraiser complex with LTS |
|---|---|
| Components |
|
-Supramolecule #1: Helraiser complex with LTS
| Supramolecule | Name: Helraiser complex with LTS / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 170 KDa |
-Macromolecule #1: Helraiser K1068Q
| Macromolecule | Name: Helraiser K1068Q / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 171.253484 KDa |
| Recombinant expression | Organism:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm) |
| Sequence | String: MSKEQLLIQR SSAAERCRRY RQKMSAEQRA SDLERRRRLQ QNVSEEQLLE KRRSEAEKQR RHRQKMSKDQ RAFEVERRRW RRQNMSREQ SSTSTTNTGR NCLLSKNGVH EDAILEHSCG GMTVRCEFCL SLNFSDEKPS DGKFTRCCSK GKVCPNDIHF P DYPAYLKR ...String: MSKEQLLIQR SSAAERCRRY RQKMSAEQRA SDLERRRRLQ QNVSEEQLLE KRRSEAEKQR RHRQKMSKDQ RAFEVERRRW RRQNMSREQ SSTSTTNTGR NCLLSKNGVH EDAILEHSCG GMTVRCEFCL SLNFSDEKPS DGKFTRCCSK GKVCPNDIHF P DYPAYLKR LMTNEDSDSK NFMENIRSIN SSFAFASMGA NIASPSGYGP YCFRIHGQVY HRTGTLHPSD GVSRKFAQLY IL DTAEATS KRLAMPENQG CSERLMININ NLMHEINELT KSYKMLHEVE KEAQSEAAAK GIAPTEVTMA IKYDRNSDPG RYN SPRVTE VAVIFRNEDG EPPFERDLLI HCKPDPNNPN ATKMKQISIL FPTLDAMTYP ILFPHGEKGW GTDIALRLRD NSVI DNNTR QNVRTRVTQM QYYGFHLSVR DTFNPILNAG KLTQQFIVDS YSKMEANRIN FIKANQSKLR VEKYSGLMDY LKSRS ENDN VPIGKMIILP SSFEGSPRNM QQRYQDAMAI VTKYGKPDLF ITMTCNPKWA DITNNLQRWQ KVENRPDLVA RVFNIK LNA LLNDICKFHL FGKVIAKIHV IEFQKRGLPH AHILLILDSE SKLRSEDDID RIVKAEIPDE DQCPRLFQIV KSNMVHG PC GIQNPNSPCM ENGKCSKGYP KEFQNATIGN IDGYPKYKRR SGSTMSIGNK VVDNTWIVPY NPYLCLKYNC HINVEVCA S IKSVKYLFKY IYKGHDCANI QISEKNIINH DEVQDFIDSR YVSAPEAVWR LFAMRMHDQS HAITRLAIHL PNDQNLYFH TDDFAEVLDR AKRHNSTLMA WFLLNREDSD ARNYYYWEIP QHYVFNNSLW TKRRKGGNKV LGRLFTVSFR EPERYYLRLL LLHVKGAIS FEDLRTVGGV TYDTFHEAAK HRGLLLDDTI WKDTIDDAII LNMPKQLRQL FAYICVFGCP SAADKLWDEN K SHFIEDFC WKLHRREGAC VNCEMHALNE IQEVFTLHGM KCSHFKLPDY PLLMNANTCD QLYEQQQAEV LINSLNDEQL AA FQTITSA IEDQTVHPKC FFLDGPGGSG QTYLYKVLTH YIRGRGGTVL PTASTGIAAN LLLGGRTFHS QYKLPIPLNE TSI SRLDIK SEVAKTIKKA QLLIIDECTM ASSHAINAID RLLREIMNLN VAFGGKVLLL GGDFRQCLSI VPHAMRSAIV QTSL KYCNV WGCFRKLSLK TNMRSEDSAY SEWLVKLGDG KLDSSFHLGM DIIEIPHEMI CNGSIIEATF GNSISIDNIK NISKR AILC PKNEHVQKLN EEILDILDGD FHTYLSDDSI DSTDDAEKEN FPIEFLNSIT PSGMPCHKLK LKVGAIIMLL RNLNSK WGL CNGTRFIIKR LRPNIIEAEV LTGSAEGEVV LIPRIDLSPS DTGLPFKLIR RQFPVMPAFA MTINKSQGQT LDRVGIF LP EPVFAHGQLY VAFSRVRRAC DVKVKVVNTS SQGKLVKHSE SVFTLNVVYR EILE |
-Macromolecule #2: LTS
| Macromolecule | Name: LTS / type: dna / ID: 2 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: Myotis lucifugus (little brown bat) |
| Molecular weight | Theoretical: 6.158058 KDa |
| Sequence | String: (DT)(DC)(DC)(DT)(DA)(DT)(DA)(DT)(DA)(DA) (DT)(DA)(DA)(DA)(DA)(DG)(DA)(DG)(DA)(DA) |
-Macromolecule #3: ZINC ION
| Macromolecule | Name: ZINC ION / type: ligand / ID: 3 / Number of copies: 1 / Formula: ZN |
|---|---|
| Molecular weight | Theoretical: 65.409 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.3 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Model: C-flat-1.2/1.3 / Material: GOLD |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Number grids imaged: 1 / Average electron dose: 73.5 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Details | The initial model has been traced manualy, then refined in Rosetta and finished in Coot 9.0 |
|---|---|
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
| Output model | PDB-7lcc: |
