 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-2286: 3D map of another peptide conjugated antibody particle by individ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-2286 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 3D map of another peptide conjugated antibody particle by individual-particle electron tomography (IPET) and optimized negative-staining. | |||||||||
 Map data Map data | Reconstruction of another IgG1 antibody by Individual-Particle Electron Microscopy (IPET). | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | peptide conjugated human IgG1 antibody | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | electron tomography / negative staining / Resolution: 16.6 Å | |||||||||
 Authors Authors | Tong H / Zhang L / Kaspar A / Rames M / Huang L / Woodnutt G / Ren G | |||||||||
 Citation Citation | Journal: Sci Rep / Year: 2013 Title: Peptide-conjugation induced conformational changes in human IgG1 observed by optimized negative-staining and individual-particle electron tomography. Authors: Huimin Tong / Lei Zhang / Allan Kaspar / Matthew J Rames / Liqing Huang / Gary Woodnutt / Gang Ren /  Abstract: Peptides show much promise as potent and selective drug candidates. Fusing peptides to a scaffold monoclonal antibody produces a conjugated antibody which has the advantages of peptide activity yet ...Peptides show much promise as potent and selective drug candidates. Fusing peptides to a scaffold monoclonal antibody produces a conjugated antibody which has the advantages of peptide activity yet also has the pharmacokinetics determined by the scaffold antibody. However, the conjugated antibody often has poor binding affinity to antigens that may be related to unknown structural changes. The study of the conformational change is difficult by conventional techniques because structural fluctuation under equilibrium results in multiple structures co-existing. Here, we employed our two recently developed electron microscopy (EM) techniques: optimized negative-staining (OpNS) EM and individual-particle electron tomography (IPET). Two-dimensional (2D) image analyses and three-dimensional (3D) maps have shown that the domains of antibodies present an elongated peptide-conjugated conformational change, suggesting that our EM techniques may be novel tools to monitor the structural conformation changes in heterogeneous and dynamic macromolecules, such as drug delivery vehicles after pharmacological synthesis and development. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera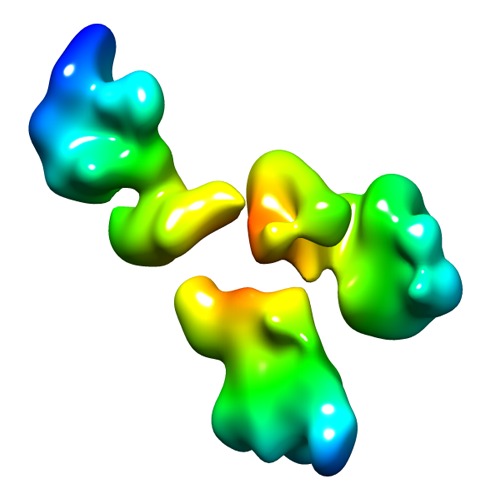
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_2286.map.gz | 17.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-2286-v30.xmlemd-2286.xml | 13.7 KB 13.7 KB | Display Display | EMDB header |
| Images | EMD-2286.tif | 145.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2286ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2286 http://ftp.pdbj.org/pub/emdb/structures/EMD-2286ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2286 | HTTPS FTP |
-Related structure data






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_2286.map.gz / Format: CCP4 / Size: 29.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of another IgG1 antibody by Individual-Particle Electron Microscopy (IPET). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.406 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Peptide-conjugated Human IgG1 Antibody
| Entire | Name: Peptide-conjugated Human IgG1 Antibody |
|---|---|
| Components |
|
-Supramolecule #1000: Peptide-conjugated Human IgG1 Antibody
| Supramolecule | Name: Peptide-conjugated Human IgG1 Antibody / type: sample / ID: 1000 Details: The sample was thawed from storage at -80 degrees Celcius before being loaded onto the grid Oligomeric state: Monomer / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 160 KDa / Theoretical: 160 KDa |
-Macromolecule #1: IgG Antibody
| Macromolecule | Name: IgG Antibody / type: protein_or_peptide / ID: 1 / Name.synonym: IgG / Number of copies: 1 / Oligomeric state: Monomer / Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / synonym: Human / Location in cell: Plasma |
| Molecular weight | Experimental: 160 KDa / Theoretical: 160 KDa |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | electron tomography |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.01 mg/mL |
|---|---|
| Buffer | pH: 7.4 Details: 1X Dulbeccos phosphate-buffered saline (Invitrogen, La Jolla, CA), 2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, and 8.1 mM Na2HPO4 |
| Staining | Type: NEGATIVE Details: EM Specimens were prepared by optimized negative-staining EM specimen preparation protocol as described Zhang L. and Ren G, Journal of Lipid Research, (2010) 51, 1228-1236; (2011) 52, 175-84 ...Details: EM Specimens were prepared by optimized negative-staining EM specimen preparation protocol as described Zhang L. and Ren G, Journal of Lipid Research, (2010) 51, 1228-1236; (2011) 52, 175-84 and BBA (2013) 1830, 2150-9. In brief, antibody was diluted to 0.01 mg/ml with deionized water. Aliquots (about 3ul) were applied to the 200 mesh glow-discharged thin carbon-coated EM grids (Cu-200CN, Pacific Grid-Tech, USA). The grid was washed by deionized water for three times, and then washed by 1% uranyl formate for three times before blotting to drying. |
| Grid | Details: 200 mesh glow-discharged thin carbon-coated EM grids (Cu-200CN, Pacific Grid-Tech, USA) |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy #1
Electron microscopy #1
| Microscopy ID | 1 |
|---|---|
| Microscope | ZEISS LIBRA120PLUS |
| Details | tilt step is 1.5 degree |
| Date | May 1, 2012 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Digitization - Sampling interval: 1.406 µm / Number real images: 95 / Average electron dose: 250 e/Å2 / Bits/pixel: 16 |
| Electron beam | Acceleration voltage: 120 kV / Electron source: LAB6 |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.2 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 80000 |
| Sample stage | Specimen holder: Gatan / Specimen holder model: OTHER / Tilt series - Axis1 - Min angle: -70.5 ° / Tilt series - Axis1 - Max angle: 70.5 ° / Tilt series - Axis1 - Angle increment: 1.5 ° |
-Electron microscopy #2
| Microscopy ID | 2 |
|---|---|
| Microscope | FEI TECNAI 20 |
| Details | tilt step is 1.5 degree |
| Date | Oct 1, 2009 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Digitization - Sampling interval: 1.406 µm / Number real images: 95 / Average electron dose: 250 e/Å2 / Bits/pixel: 16 |
| Electron beam | Acceleration voltage: 200 kV / Electron source: LAB6 |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 80000 |
| Sample stage | Specimen holder: Gatan / Specimen holder model: OTHER / Tilt series - Axis1 - Min angle: -70.5 ° / Tilt series - Axis1 - Max angle: 70.5 ° / Tilt series - Axis1 - Angle increment: 1.5 ° |
-Image processing
| Details | Reconstruction was done by Individual-Particle Tomography Reconstruction (IPET) method.{eulerAnglesDetails}: Tomography tilt angle from -70.5 to 70.5 in step of 1.5 |
|---|---|
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 16.6 Å / Resolution method: OTHER Software - Name: Individual-partcile, electron, tomogrphy, (IPET), and, FETR Details: Map was reconstructed by individual-particle electron tomography (IPET)and Focus ET Reconstruction Algorithm. Number images used: 95 |
| CTF correction | Details: TOMOCTF |
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Software | Name: Manual |
| Details | Manually fit each domain into the density map in order to compare the conformational different between the peptide-conjugated antibody structure and PDB structure in term of domain size and shape. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |

