 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
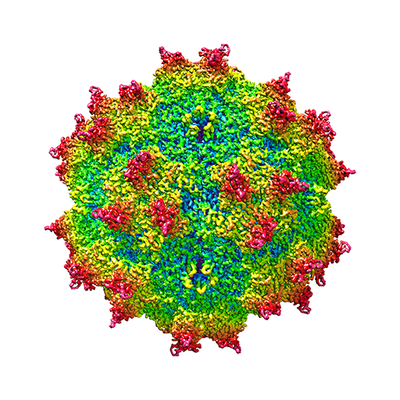
Yorodumi- EMDB-22854: Adeno-Associated Virus (AAV-DJ) - cryo-EM structure at 1.56 Angst... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22854 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Adeno-Associated Virus (AAV-DJ) - cryo-EM structure at 1.56 Angstrom Resolution | |||||||||
 Map data Map data | Full particle Relion map, sharpened using volume-whitening routine of cisTEM. Cropped version also provided for ease of comparison with coordinates. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Gene therapy vector / VIRUS LIKE PARTICLE / VIRUS | |||||||||
| Function / homology |  Function and homology information Function and homology informationT=1 icosahedral viral capsid / structural molecule activity / metal ion binding Similarity search - Function | |||||||||
| Biological species |   Adeno-associated virus Adeno-associated virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 1.56 Å | |||||||||
 Authors Authors | Xie Q / Yoshioka CK | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Viruses / Year: 2020 Title: Adeno-Associated Virus (AAV-DJ)-Cryo-EM Structure at 1.56 Å Resolution. Authors: Qing Xie / Craig K Yoshioka / Michael S Chapman / Abstract: Adeno-associated virus is the leading viral vector for gene therapy. AAV-DJ is a recombinant variant developed for tropism to the liver. The AAV-DJ structure has been determined to 1.56 Å resolution ...Adeno-associated virus is the leading viral vector for gene therapy. AAV-DJ is a recombinant variant developed for tropism to the liver. The AAV-DJ structure has been determined to 1.56 Å resolution through cryo-electron microscopy (cryo-EM). Only apoferritin is reported in preprints at 1.6 Å or higher resolution, and AAV-DJ nearly matches the highest resolutions ever attained through X-ray diffraction of virus crystals. However, cryo-EM has the advantage that most of the hydrogens are clear, improving the accuracy of atomic refinement, and removing ambiguity in hydrogen bond identification. Outside of secondary structures where hydrogen bonding was predictable a priori, the networks of hydrogen bonds coming from direct observation of hydrogens and acceptor atoms are quite different from those inferred even at 2.8 Å resolution. The implications for understanding viral assembly mean that cryo-EM will likely become the favored approach for high resolution structural virology. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22854.map.gz | 765.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22854-v30.xmlemd-22854.xml | 28.1 KB 28.1 KB | Display Display | EMDB header |
| Images |  emd_22854.png emd_22854.png | 187.6 KB | ||
| Masks | emd_22854_msk_1.map | 824 MB | Mask map | |
| Filedesc metadata | emd-22854.cif.gz | 7.7 KB | ||
| Others | emd_22854_additional_1.map.gzemd_22854_additional_2.map.gzemd_22854_additional_3.map.gzemd_22854_half_map_1.map.gzemd_22854_half_map_2.map.gz | 752.3 MB 33.8 MB 34.7 MB 764.8 MB 764.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22854ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22854 http://ftp.pdbj.org/pub/emdb/structures/EMD-22854ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22854 | HTTPS FTP |
-Related structure data
| Related structure data |  7kfrMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10551 (Title: Adeno-Associated Virus (AAV-DJ)-Cryo-EM Structure at 1.56 Å Resolution Data size: 2.8 TB Data #1: Falcon3 frames converted to TIFF and compressed [micrographs - multiframe]) |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_22854.map.gz / Format: CCP4 / Size: 824 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Full particle Relion map, sharpened using volume-whitening routine of cisTEM. Cropped version also provided for ease of comparison with coordinates. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.5105 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) X (Row.)
X (Row.) Y (Col.)
Y (Col.)





















-Supplemental data
-Mask #1
| File | emd_22854_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
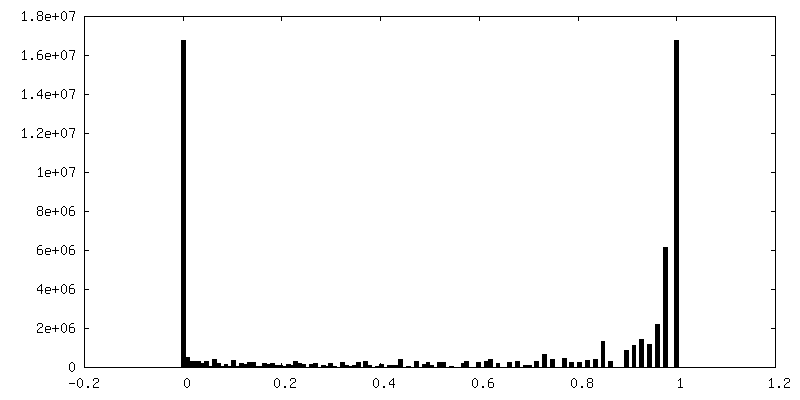
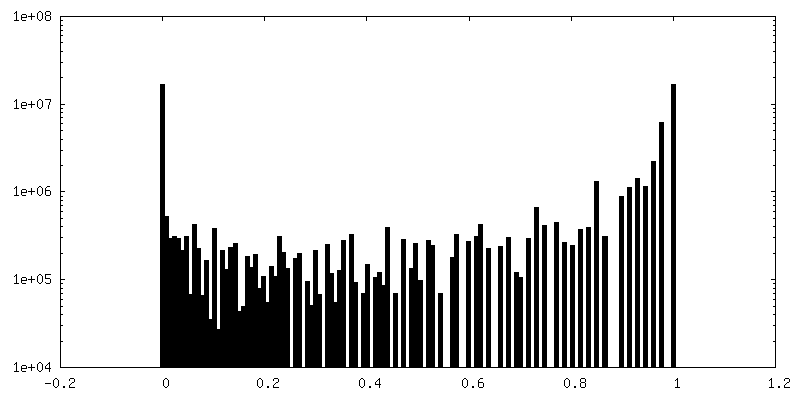
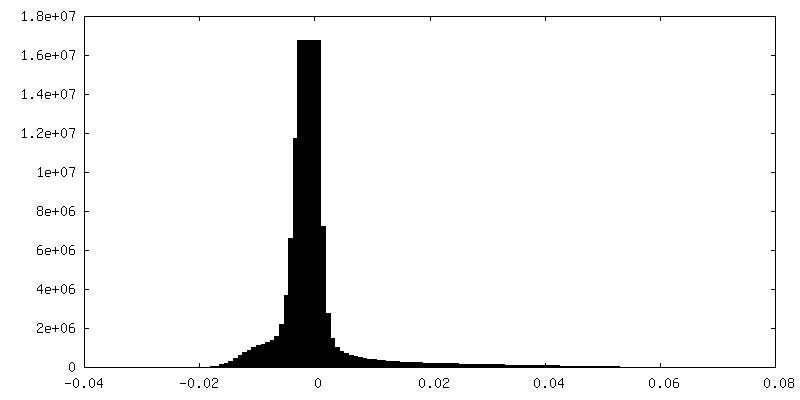
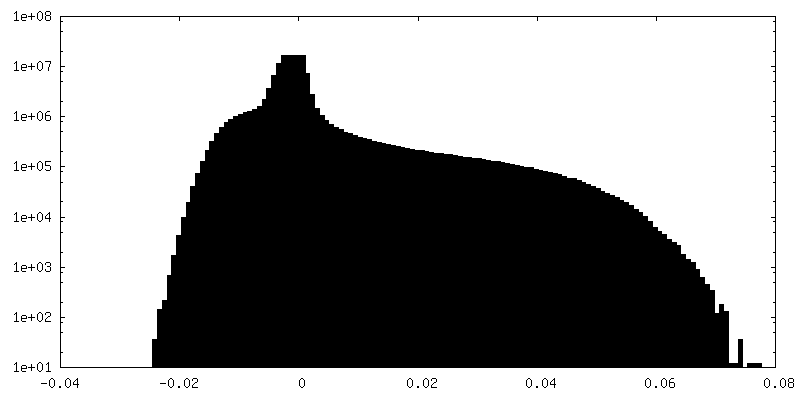
| Density Histograms |






-Additional map: Full particle, unsharpened Relion map, low pass filtered...
| File | emd_22854_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Full particle, unsharpened Relion map, low pass filtered beyond FSC of 1.56 A. See also cropped version. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



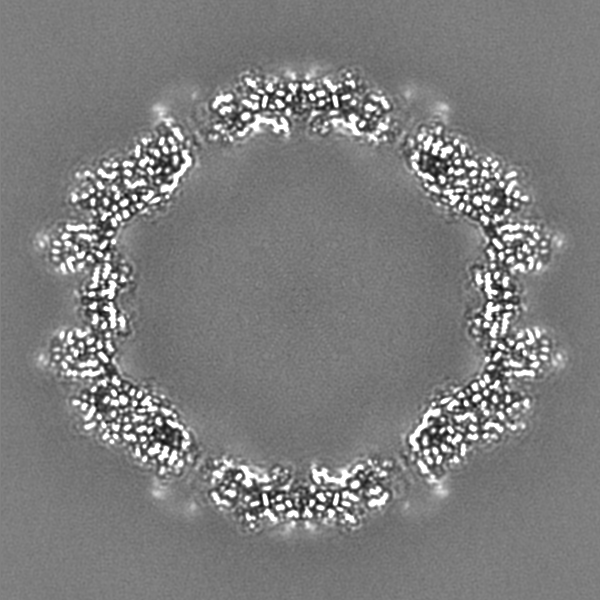
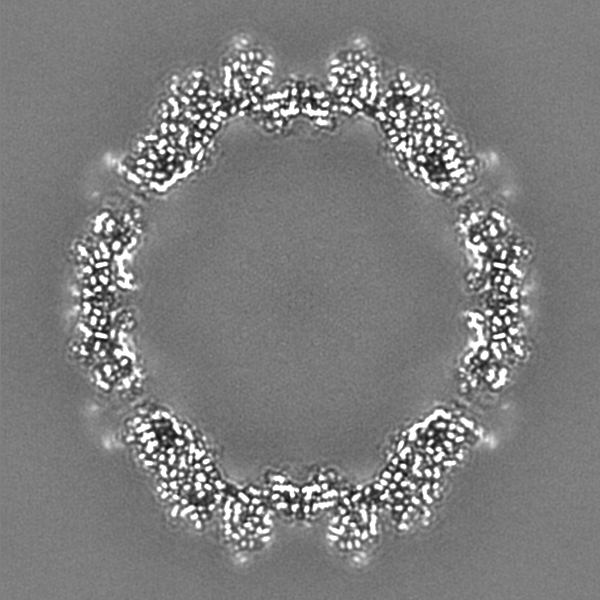
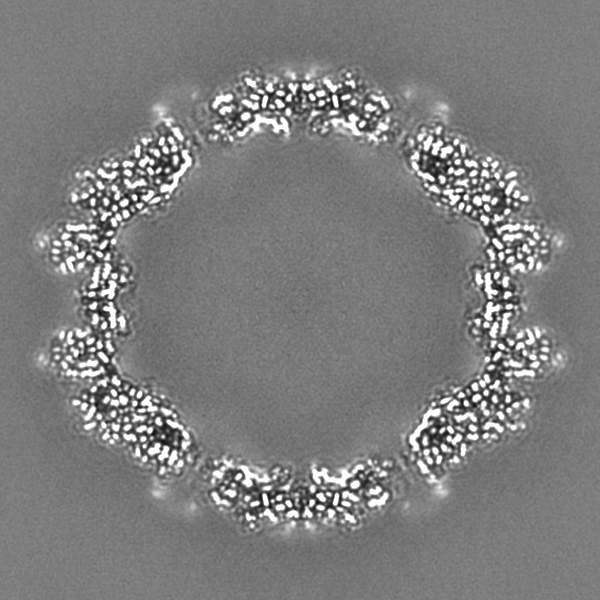

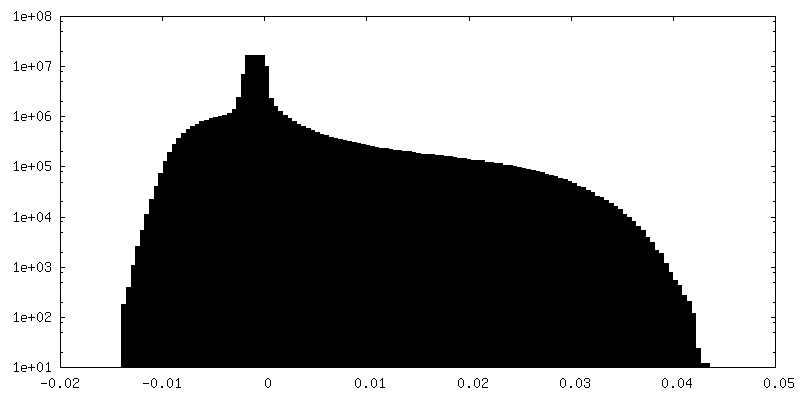
-Additional map: Unsharpened Relion map, cropped ~20 Angstrom outside subunit...
| File | emd_22854_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened Relion map, cropped ~20 Angstrom outside subunit coordinates deposited, for ease of visualization. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

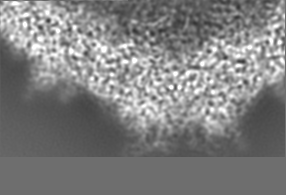
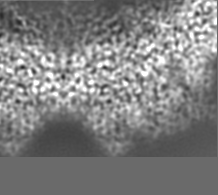
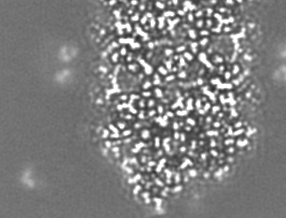
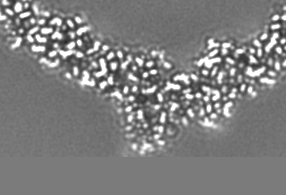
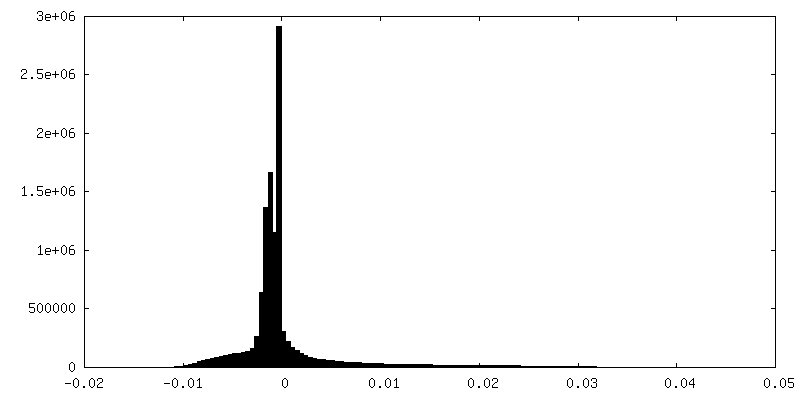
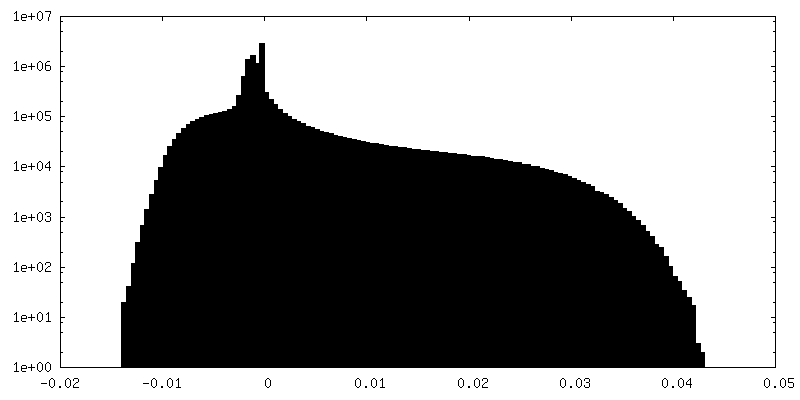
-Additional map: Relion map, sharpened using volume-whitening routine of cisTEM,...
| File | emd_22854_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion map, sharpened using volume-whitening routine of cisTEM, and cropped ~20 Angstrom outside deposited subunit coordinates for ease of comparison. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



-Half map: Relion half map 1.
| File | emd_22854_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion half map 1. | ||||||||||||
| Projections & Slices |
| ||||||||||||
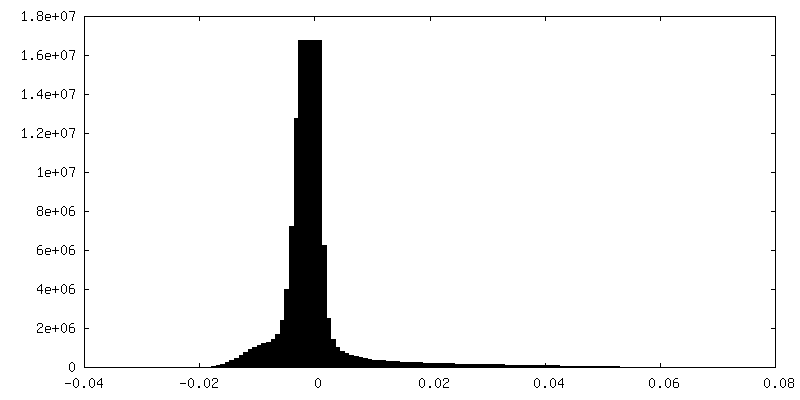
| Density Histograms |



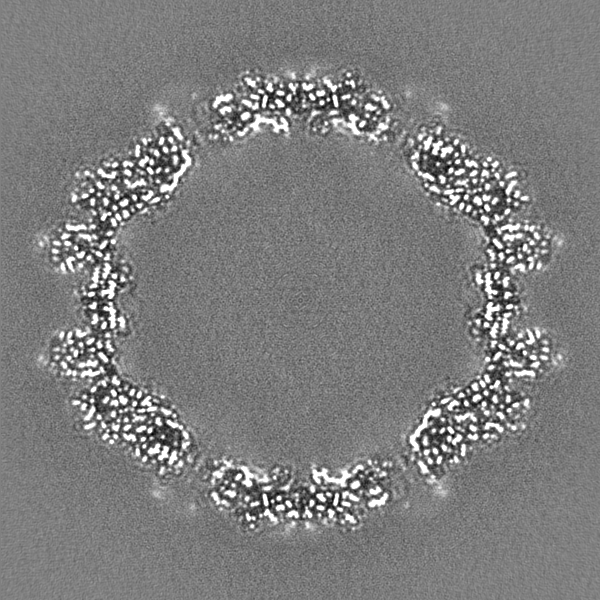
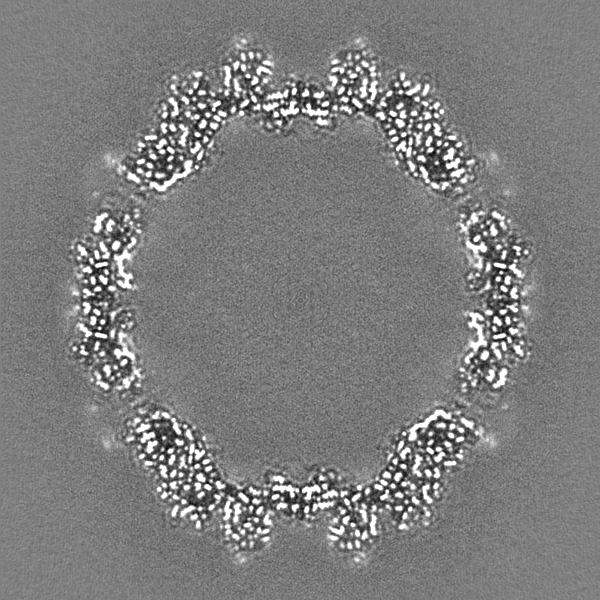
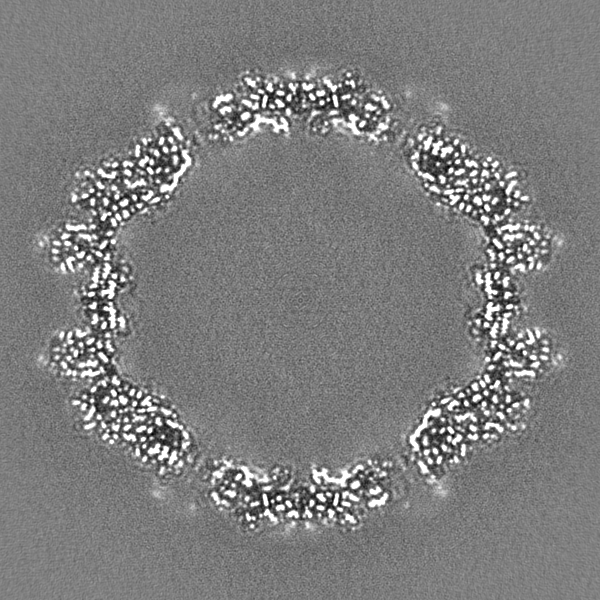
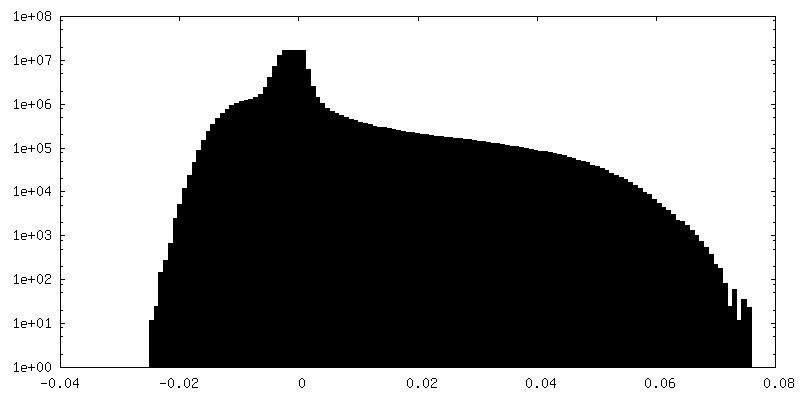
-Half map: Relion half map 2.
| File | emd_22854_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Relion half map 2. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



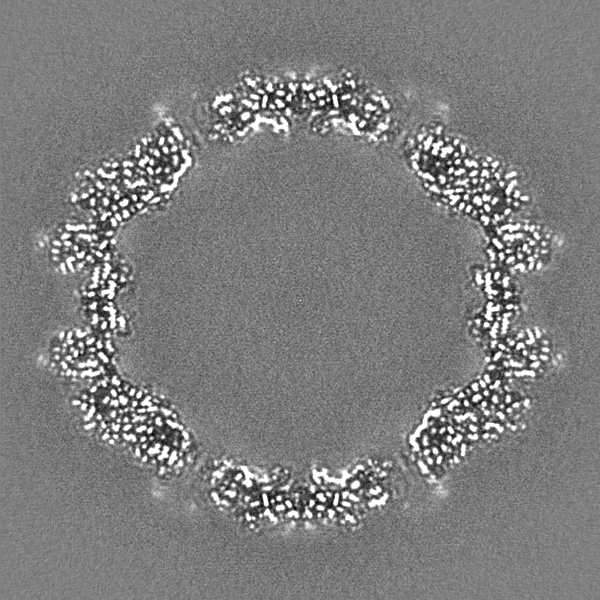
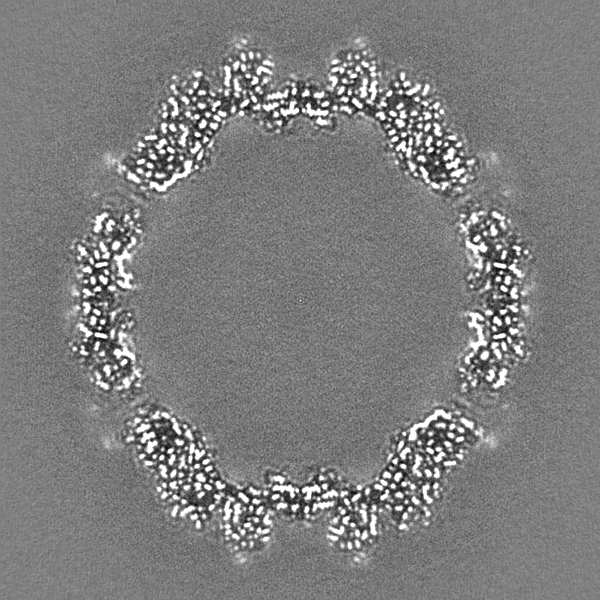
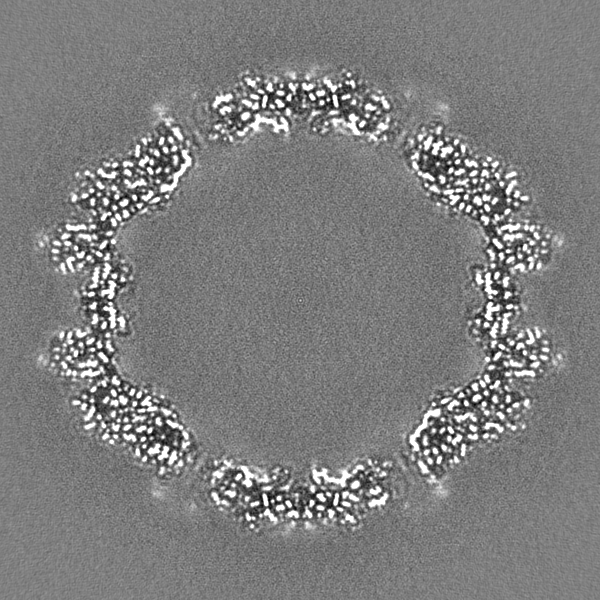
- Sample components
Sample components
-Entire : Adeno-associated virus
| Entire | Name: Adeno-associated virus |
|---|---|
| Components |
|
-Supramolecule #1: Adeno-associated virus
| Supramolecule | Name: Adeno-associated virus / type: virus / ID: 1 / Parent: 0 / Macromolecule list: #1 / NCBI-ID: 272636 / Sci species name: Adeno-associated virus / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: STRAIN / Virus enveloped: No / Virus empty: Yes |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Molecular weight | Theoretical: 3.746 MDa |
| Virus shell | Shell ID: 1 / Diameter: 250.0 Å / T number (triangulation number): 1 |
-Macromolecule #1: Capsid protein VP1
| Macromolecule | Name: Capsid protein VP1 / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Adeno-associated virus |
| Molecular weight | Theoretical: 58.877809 KDa |
| Recombinant expression | Organism:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm) |
| Sequence | String: GADGVGNSSG NWHCDSTWMG DRVITTSTRT WALPTYNNHL YKQISNSTSG GSSNDNAYFG YSTPWGYFDF NRFHCHFSPR DWQRLINNN WGFRPKRLSF KLFNIQVKEV TQNEGTKTIA NNLTSTIQVF TDSEYQLPYV LGSAHQGCLP PFPADVFMIP Q YGYLTLNN ...String: GADGVGNSSG NWHCDSTWMG DRVITTSTRT WALPTYNNHL YKQISNSTSG GSSNDNAYFG YSTPWGYFDF NRFHCHFSPR DWQRLINNN WGFRPKRLSF KLFNIQVKEV TQNEGTKTIA NNLTSTIQVF TDSEYQLPYV LGSAHQGCLP PFPADVFMIP Q YGYLTLNN GSQAVGRSSF YCLEYFPSQM LRTGNNFQFT YTFEDVPFHS SYAHSQSLDR LMNPLIDQYL YYLSRTQTTG GT TNTQTLG FSQGGPNTMA NQAKNWLPGP CYRQQRVSKT SADNNNSEYS WTGATKYHLN GRDSLVNPGP AMASHKDDEE KFF PQSGVL IFGKQGSEKT NVDIEKVMIT DEEEIRTTNP VATEQYGSVS TNLQRGNRQA ATADVNTQGV LPGMVWQDRD VYLQ GPIWA KIPHTDGHFH PSPLMGGFGL KHPPPQILIK NTPVPADPPT TFNQSKLNSF ITQYSTGQVS VEIEWELQKE NSKRW NPEI QYTSNYYKST SVDFAVNTEG VYSEPRPIGT RYLTRNL UniProtKB: Capsid protein VP1 |
-Macromolecule #2: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 2 / Number of copies: 3 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 265 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.6 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
| ||||||||||||
| Grid | Model: Quantifoil / Material: COPPER / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: OTHER / Pretreatment - Pressure: 0.03 kPa Details: PELCO easiGlow Glow Discharge Cleaning System Current: 15mA | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 293 K / Instrument: FEI VITROBOT MARK IV Details: Two 3uL aliquots applied to grid (manual blotting between), prior to automated 3 second blot before plunging.. | ||||||||||||
| Details | Monodisperse |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 93.0 K / Max: 93.0 K |
| Details | Coma-free alignment and objective astigmatism where corrected using Sherpa (Thermo Fisher, Inc.). |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: INTEGRATING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 2241 / Average exposure time: 15.8 sec. / Average electron dose: 30.0 e/Å2 Details: Data were collected with pixel size of 0.514 angstrom on a FEI Titan Krios (Thermo Fisher, Inc.) at 300 kV, using a Falcon 3 camera (Thermo Fisher) with a total dose of approx. 30 e-/A2 ...Details: Data were collected with pixel size of 0.514 angstrom on a FEI Titan Krios (Thermo Fisher, Inc.) at 300 kV, using a Falcon 3 camera (Thermo Fisher) with a total dose of approx. 30 e-/A2 fractionated across 200 frames. Camera dose rate was approx. 0.5 e-/pixel/s. Defocus was random in the nominal range of -0.8 to -2.6 um. Images were acquired in EPU (Thermo Fisher, Inc.) without using image shift. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 154400 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: -2.6 µm / Nominal defocus min: -0.8 µm / Nominal magnification: 155000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | Stand-alone RSRef was used for refinement of magnification, resolution, envelope correction and atomic B-factors. This was alternated with RSRef-embedded CNS was used for molecular dynamics optimization (1st round) and stereochemically-restrained all-atom least-squares optimization. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Least-squares residual |
| Output model | PDB-7kfr: |

