 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-21371: Single particle reconstruction of glucose isomerase from Streptom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-21371 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Single particle reconstruction of glucose isomerase from Streptomyces rubiginosus based on data acquired in the presence of substantial aberrations | ||||||||||||||||||
 Map data Map data | Single particle reconstruction of glucose isomerase from Streptomyces rubiginosus based on data acquired in the presence of substantial aberrations | ||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | glucose isomerase / ISOMERASE | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationxylose isomerase / xylose isomerase activity / D-xylose metabolic process / magnesium ion binding / identical protein binding / cytoplasm Similarity search - Function | ||||||||||||||||||
| Biological species |  Streptomyces rubiginosus (bacteria) Streptomyces rubiginosus (bacteria) | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.7 Å | ||||||||||||||||||
 Authors Authors | Bromberg R / Guo Y | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: IUCrJ / Year: 2020 Title: High-resolution cryo-EM reconstructions in the presence of substantial aberrations. Authors: Raquel Bromberg / Yirui Guo / Dominika Borek / Zbyszek Otwinowski / Abstract: Here, an analysis is performed of how uncorrected antisymmetric aberrations, such as coma and trefoil, affect cryo-EM single-particle reconstruction (SPR) results, and an analytical formula ...Here, an analysis is performed of how uncorrected antisymmetric aberrations, such as coma and trefoil, affect cryo-EM single-particle reconstruction (SPR) results, and an analytical formula quantifying information loss owing to their presence is inferred that explains why Fourier-shell coefficient-based statistics may report significantly overestimated resolution if these aberrations are not fully corrected. The analysis is validated with reference-based aberration refinement for two cryo-EM SPR data sets acquired with a 200 kV microscope in the presence of coma exceeding 40 µm, and 2.3 and 2.7 Å reconstructions for 144 and 173 kDa particles, respectively, were obtained. The results provide a description of an efficient approach for assessing information loss in cryo-EM SPR data acquired in the presence of higher order aberrations, and address inconsistent guidelines regarding the level of aberrations that is acceptable in cryo-EM SPR experiments. #1: Journal: Biorxiv / Year: 2020Title: High-resolution cryo-EM reconstructions in the presence of substantial aberrations Authors: Bromberg R / Guo Y / Borek D / Otwinowski Z | ||||||||||||||||||
| History |
|
- Structure visualization
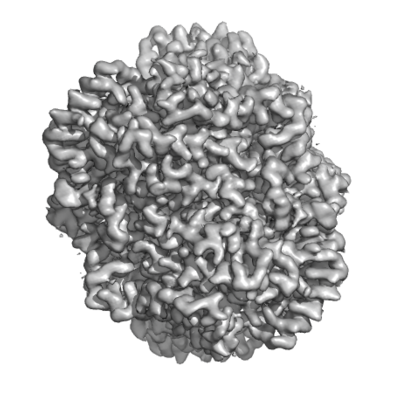
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_21371.map.gz | 1.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-21371-v30.xmlemd-21371.xml | 15.6 KB 15.6 KB | Display Display | EMDB header |
| Images |  emd_21371.png emd_21371.png | 148.9 KB | ||
| Filedesc metadata | emd-21371.cif.gz | 5.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-21371ftp://ftp.pdbj.org/pub/emdb/structures/EMD-21371 http://ftp.pdbj.org/pub/emdb/structures/EMD-21371ftp://ftp.pdbj.org/pub/emdb/structures/EMD-21371 | HTTPS FTP |
-Related structure data
| Related structure data |  6vrsMC  6vsaC  6vscC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10360 (Title: 2.7 Angstrom cryo-EM reconstructions of glucose isomerase in the presence of substantial aberrations Data size: 1.0 TB Data #1: Unaligned multiframe data for xylose isomerase (glucose isomerase) [micrographs - multiframe]) |












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_21371.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Single particle reconstruction of glucose isomerase from Streptomyces rubiginosus based on data acquired in the presence of substantial aberrations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.91 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : glucose isomerase
| Entire | Name: glucose isomerase |
|---|---|
| Components |
|
-Supramolecule #1: glucose isomerase
| Supramolecule | Name: glucose isomerase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Streptomyces rubiginosus (bacteria) |
| Molecular weight | Theoretical: 172.9 KDa |
-Macromolecule #1: xylose isomerase
| Macromolecule | Name: xylose isomerase / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO / EC number: xylose isomerase |
|---|---|
| Source (natural) | Organism: Streptomyces rubiginosus (bacteria) |
| Molecular weight | Theoretical: 43.283297 KDa |
| Sequence | String: MNYQPTPEDR FTFGLWTVGW QGRDPFGDAT RRALDPVESV RRLAELGAHG VTFHDDDLIP FGSSDSEREE HVKRFRQALD DTGMKVPMA TTNLFTHPVF KDGGFTANDR DVRRYALRKT IRNIDLAVEL GAETYVAWGG REGAESGGAK DVRDALDRMK E AFDLLGEY ...String: MNYQPTPEDR FTFGLWTVGW QGRDPFGDAT RRALDPVESV RRLAELGAHG VTFHDDDLIP FGSSDSEREE HVKRFRQALD DTGMKVPMA TTNLFTHPVF KDGGFTANDR DVRRYALRKT IRNIDLAVEL GAETYVAWGG REGAESGGAK DVRDALDRMK E AFDLLGEY VTSQGYDIRF AIEPKPNEPR GDILLPTVGH ALAFIERLER PELYGVNPEV GHEQMAGLNF PHGIAQALWA GK LFHIDLN GQNGIKYDQD LRFGAGDLRA AFWLVDLLES AGYSGPRHFD FKPPRTEDFD GVWASAAGCM RNYLILKERA AAF RADPEV QEALRASRLD ELARPTAADG LQALLDDRSA FEEFDVDAAA ARGMAFERLD QLAMDHLLGA RG UniProtKB: Xylose isomerase |
-Macromolecule #2: MANGANESE (II) ION
| Macromolecule | Name: MANGANESE (II) ION / type: ligand / ID: 2 / Number of copies: 8 / Formula: MN |
|---|---|
| Molecular weight | Theoretical: 54.938 Da |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 16 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 40 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Component - Concentration: 20.0 mM / Component - Name: HEPES |
| Grid | Model: Quantifoil R1.2/1.3 / Material: GOLD / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS TALOS |
|---|---|
| Alignment procedure | Basic - Residual tilt: 5.3 mrad |
| Details | The goal of the experiment was to show that it is possible to perform high resolution reconstruction in the presence of higher order aberrations. |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-200 / Number grids imaged: 1 / Number real images: 202 / Average exposure time: 100.0 sec. / Average electron dose: 120.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.0 µm |
| Sample stage | Cooling holder cryogen: NITROGEN |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | COOT was crucial as well. |
| Refinement | Space: RECIPROCAL / Protocol: OTHER / Target criteria: REFMAC |
| Output model | PDB-6vrs: |

