Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-20962: Cryo-EM structure of nucleotide-free MsbA reconstituted into pept... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-20962 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
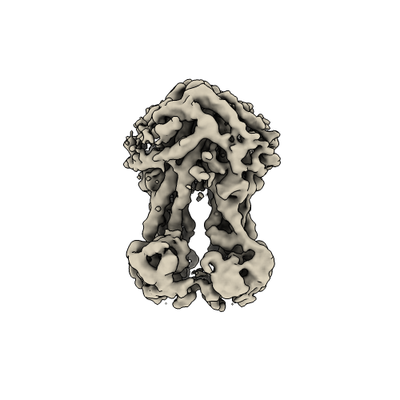
| Title | Cryo-EM structure of nucleotide-free MsbA reconstituted into peptidiscs, conformation 2 | |||||||||
 Map data Map data | nucleotide-free MsbA reconstituted into peptidiscs, conformation 2 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | membrane protein / ABC transporter / membrane mimetic / peptidisc / TRANSLOCASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationMsbA transporter complex / lipopolysaccharide floppase activity / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to catalyse transmembrane movement of substances / lipid translocation / ABC-type lipid A-core oligosaccharide transporter / ABC-type oligopeptide transporter activity / lipopolysaccharide transport / ATPase-coupled lipid transmembrane transporter activity / ABC-type xenobiotic transporter activity / lipid transport ...MsbA transporter complex / lipopolysaccharide floppase activity / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to catalyse transmembrane movement of substances / lipid translocation / ABC-type lipid A-core oligosaccharide transporter / ABC-type oligopeptide transporter activity / lipopolysaccharide transport / ATPase-coupled lipid transmembrane transporter activity / ABC-type xenobiotic transporter activity / lipid transport / ATP-binding cassette (ABC) transporter complex / transmembrane transport / lipid binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
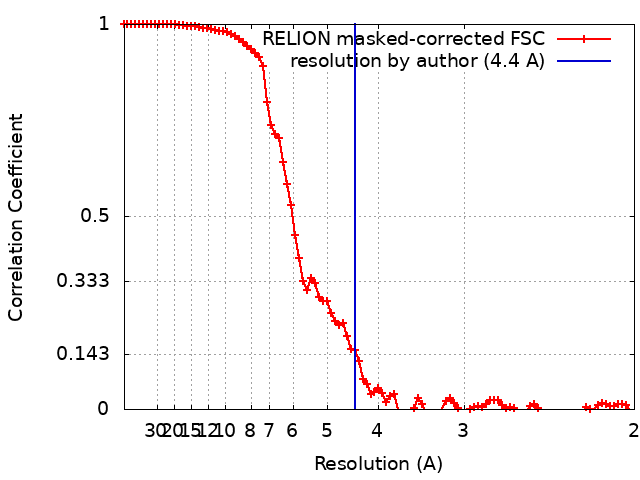
| Method | single particle reconstruction / cryo EM / Resolution: 4.4 Å | |||||||||
 Authors Authors | Angiulli G / Walz T | |||||||||
 Citation Citation | Journal: Elife / Year: 2020 Title: New approach for membrane protein reconstitution into peptidiscs and basis for their adaptability to different proteins. Authors: Gabriella Angiulli / Harveer Singh Dhupar / Hiroshi Suzuki / Irvinder Singh Wason / Franck Duong Van Hoa / Thomas Walz /   Abstract: Previously we introduced peptidiscs as an alternative to detergents to stabilize membrane proteins in solution (Carlson et al., 2018). Here, we present 'on-gradient' reconstitution, a new gentle ...Previously we introduced peptidiscs as an alternative to detergents to stabilize membrane proteins in solution (Carlson et al., 2018). Here, we present 'on-gradient' reconstitution, a new gentle approach for the reconstitution of labile membrane-protein complexes, and used it to reconstitute reaction center complexes, demonstrating that peptidiscs can adapt to transmembrane domains of very different sizes and shapes. Using the conventional 'on-bead' approach, we reconstituted proteins MsbA and MscS and find that peptidiscs stabilize them in their native conformation and allow for high-resolution structure determination by cryo-electron microscopy. The structures reveal that peptidisc peptides can arrange around transmembrane proteins differently, thus revealing the structural basis for why peptidiscs can stabilize such a large variety of membrane proteins. Together, our results establish the gentle and easy-to-use peptidiscs as a potentially universal alternative to detergents as a means to stabilize membrane proteins in solution for structural and functional studies. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_20962.map.gz | 59.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-20962-v30.xmlemd-20962.xml | 16.6 KB 16.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_20962_fsc.xml | 9.2 KB | Display | FSC data file |
| Images |  emd_20962.png emd_20962.png | 67.2 KB | ||
| Filedesc metadata | emd-20962.cif.gz | 6.4 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20962ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20962 http://ftp.pdbj.org/pub/emdb/structures/EMD-20962ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20962 | HTTPS FTP |
-Related structure data
| Related structure data |  6uzlMC  6uz2C  6uzhC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_20962.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | nucleotide-free MsbA reconstituted into peptidiscs, conformation 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Lipid A export ATP-binding/permease protein MsbA
| Entire | Name: Lipid A export ATP-binding/permease protein MsbA |
|---|---|
| Components |
|
-Supramolecule #1: Lipid A export ATP-binding/permease protein MsbA
| Supramolecule | Name: Lipid A export ATP-binding/permease protein MsbA / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Lipid A export ATP-binding/permease protein MsbA
| Macromolecule | Name: Lipid A export ATP-binding/permease protein MsbA / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO EC number: ABC-type lipid A-core oligosaccharide transporter |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 66.833109 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MGSSHHHHHH SSGLVPRGSH MMHNDKDLST WQTFRRLWPT IAPFKAGLIV AGVALILNAA SDTFMLSLLK PLLDDGFGKT DRSVLVWMP LVVIGLMILR GITSYVSSYC ISWVSGKVVM TMRRRLFGHM MGMPVSFFDK QSTGTLLSRI TYDSEQVASS S SGALITVV ...String: MGSSHHHHHH SSGLVPRGSH MMHNDKDLST WQTFRRLWPT IAPFKAGLIV AGVALILNAA SDTFMLSLLK PLLDDGFGKT DRSVLVWMP LVVIGLMILR GITSYVSSYC ISWVSGKVVM TMRRRLFGHM MGMPVSFFDK QSTGTLLSRI TYDSEQVASS S SGALITVV REGASIIGLF IMMFYYSWQL SIILIVLAPI VSIAIRVVSK RFRNISKNMQ NTMGQVTTSA EQMLKGHKEV LI FGGQEVE TKRFDKVSNR MRLQGMKMVS ASSISDPIIQ LIASLALAFV LYAASFPSVM DSLTAGTITV VFSSMIALMR PLK SLTNVN AQFQRGMAAC QTLFTILDSE QEKDEGKRVI ERATGDVEFR NVTFTYPGRD VPALRNINLK IPAGKTVALV GRSG SGKST IASLITRFYD IDEGEILMDG HDLREYTLAS LRNQVALVSQ NVHLFNDTVA NNIAYARTEQ YSREQIEEAA RMAYA MDFI NKMDNGLDTV IGENGVLLSG GQRQRIAIAR ALLRDSPILI LDEATSALDT ESERAIQAAL DELQKNRTSL VIAHRL STI EKADEIVVVE DGVIVERGTH NDLLEHRGVY AQLHKMQFGQ UniProtKB: ATP-binding transport protein MsbA |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.3 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.9 Component:
| |||||||||
| Sugar embedding | Material: vitreous ice | |||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 40 sec. / Pretreatment - Atmosphere: AIR | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Number grids imaged: 2 / Average exposure time: 10.0 sec. / Average electron dose: 80.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: -2.0 µm / Nominal defocus min: -1.0 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |