 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-11792 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | MutS in mismatch bound state | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | DNA Mismatch Repair MutS / DNA BINDING PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationadenine/cytosine mispair binding / MutS complex / mismatch repair complex / regulation of DNA recombination / mismatched DNA binding / DNA binding, bending / ATP-dependent DNA damage sensor activity / mismatch repair / ADP binding / damaged DNA binding ...adenine/cytosine mispair binding / MutS complex / mismatch repair complex / regulation of DNA recombination / mismatched DNA binding / DNA binding, bending / ATP-dependent DNA damage sensor activity / mismatch repair / ADP binding / damaged DNA binding / DNA damage response / ATP hydrolysis activity / ATP binding / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.9 Å | |||||||||
 Authors Authors | Fernandez-Leiro R / Bhairosing-Kok D | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2021 Title: The selection process of licensing a DNA mismatch for repair. Authors: Rafael Fernandez-Leiro / Doreth Bhairosing-Kok / Vladislav Kunetsky / Charlie Laffeber / Herrie H Winterwerp / Flora Groothuizen / Alexander Fish / Joyce H G Lebbink / Peter Friedhoff / ...Authors: Rafael Fernandez-Leiro / Doreth Bhairosing-Kok / Vladislav Kunetsky / Charlie Laffeber / Herrie H Winterwerp / Flora Groothuizen / Alexander Fish / Joyce H G Lebbink / Peter Friedhoff / Titia K Sixma / Meindert H Lamers /    Abstract: DNA mismatch repair detects and removes mismatches from DNA by a conserved mechanism, reducing the error rate of DNA replication by 100- to 1,000-fold. In this process, MutS homologs scan DNA, ...DNA mismatch repair detects and removes mismatches from DNA by a conserved mechanism, reducing the error rate of DNA replication by 100- to 1,000-fold. In this process, MutS homologs scan DNA, recognize mismatches and initiate repair. How the MutS homologs selectively license repair of a mismatch among millions of matched base pairs is not understood. Here we present four cryo-EM structures of Escherichia coli MutS that provide snapshots, from scanning homoduplex DNA to mismatch binding and MutL activation via an intermediate state. During scanning, the homoduplex DNA forms a steric block that prevents MutS from transitioning into the MutL-bound clamp state, which can only be overcome through kinking of the DNA at a mismatch. Structural asymmetry in all four structures indicates a division of labor between the two MutS monomers. Together, these structures reveal how a small conformational change from the homoduplex- to heteroduplex-bound MutS acts as a licensing step that triggers a dramatic conformational change that enables MutL binding and initiation of the repair cascade. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_11792.map.gz | 12.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-11792-v30.xmlemd-11792.xml | 22.5 KB 22.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_11792_fsc.xml | 7 KB | Display | FSC data file |
| Images | 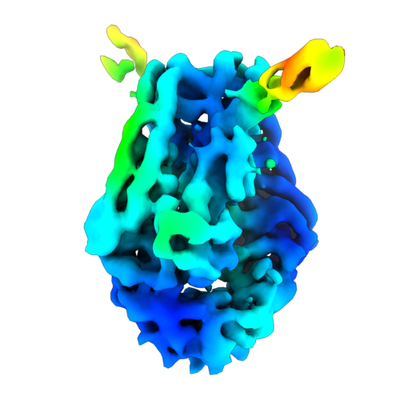 emd_11792.png emd_11792.png | 91.3 KB | ||
| Masks | emd_11792_msk_1.map | 27 MB | Mask map | |
| Filedesc metadata | emd-11792.cif.gz | 6.9 KB | ||
| Others | emd_11792_half_map_1.map.gzemd_11792_half_map_2.map.gz | 20.7 MB 20.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-11792ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11792 http://ftp.pdbj.org/pub/emdb/structures/EMD-11792ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11792 | HTTPS FTP |
-Related structure data
| Related structure data |  7ai6MC  7ai5C  7ai7C  7aibC  7aicC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_11792.map.gz / Format: CCP4 / Size: 27 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.76 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_11792_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_11792_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_11792_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : MutS loaded on DNA substrate with one central mismatched basepair...
| Entire | Name: MutS loaded on DNA substrate with one central mismatched basepair in the presence of AMP-PNP |
|---|---|
| Components |
|
-Supramolecule #1: MutS loaded on DNA substrate with one central mismatched basepair...
| Supramolecule | Name: MutS loaded on DNA substrate with one central mismatched basepair in the presence of AMP-PNP type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#3 |
|---|---|
| Molecular weight | Theoretical: 190 KDa |
-Supramolecule #2: DNA mismatch repair protein MutS
| Supramolecule | Name: DNA mismatch repair protein MutS / type: complex / ID: 2 / Parent: 1 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
-Supramolecule #3: DNA
| Supramolecule | Name: DNA / type: complex / ID: 3 / Parent: 1 / Macromolecule list: #2-#3 |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
-Macromolecule #1: DNA mismatch repair protein MutS
| Macromolecule | Name: DNA mismatch repair protein MutS / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 95.269625 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MSAIENFDAH TPMMQQYLRL KAQHPEILLF YRMGDFYELF YDDAKRASQL LDISLTKRGA SAGEPIPMAG IPYHAVENYL AKLVNQGES VAIAEQIGDP ATSKGPVERK VVRIVTPGTI SDEALLQERQ DNLLAAIWQD SKGFGYATLD ISSGRFRLSE P ADRETMAA ...String: MSAIENFDAH TPMMQQYLRL KAQHPEILLF YRMGDFYELF YDDAKRASQL LDISLTKRGA SAGEPIPMAG IPYHAVENYL AKLVNQGES VAIAEQIGDP ATSKGPVERK VVRIVTPGTI SDEALLQERQ DNLLAAIWQD SKGFGYATLD ISSGRFRLSE P ADRETMAA ELQRTNPAEL LYAEDFAEMS LIEGRRGLRR RPLWEFEIDT ARQQLNLQFG TRDLVGFGVE NAPRGLSAAG AL LQYAKCT QRTTLPHIRS ITMEREQDSI IMDAATRRNL EITQNLAGGA ENTLASVLDS TVTPMGSRML KRWLHMPVRD TRV LLERQQ TIGALQDFTA GLQPVLRQVG DLERILARLA LRTARPRDLA RMRHAFQQLP ELRAQLETVD SAPVQALREK MGEF AELRD LLERAIIDTP PVLVRDGGVI ASGYNEELDE WRALADGATD YLERLEVRER ERTGLDTLKV GFNAVHGYYI QISRG QSHL APINYMRRQT LKNAERYIIP ELKEYEDKVL TSKGKALALE KQLYEELFDL LLPHLEALQQ SASALAELDV LVNLAE RAY TLNYTSPTFI DKPGIRITEG RHPVVEQVLN EPFIANPLNL SPQRRMLIIT GPNMGGKSTY MRQTALIALM AYIGSYV PA QKVEIGPIDR IFTRVGAADD LASGRSTFMV EMTETANILH NATEYSLVLM DEIGRGTSTY DGLSLAWAVA ENLANKIK A LTLFATHYFE LTQLPEKMEG VANVHLDALE HGDTIAFMHS VQDGAASKSY GLAVAALAGV PKEVIKRARQ KLRELESIS PNAAATQVDG TQMSLLSVPE ETSPAVEALE NLDPRSLTPR QALEWIYRLK SLV UniProtKB: DNA mismatch repair protein MutS |
-Macromolecule #2: DNA (25-MER)
| Macromolecule | Name: DNA (25-MER) / type: dna / ID: 2 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 7.780007 KDa |
| Sequence | String: (DG)(DG)(DA)(DT)(DC)(DA)(DT)(DC)(DG)(DA) (DG)(DG)(DA)(DT)(DC)(DG)(DA)(DG)(DC)(DT) (DC)(DG)(DG)(DT)(DG) |
-Macromolecule #3: DNA (25-MER)
| Macromolecule | Name: DNA (25-MER) / type: dna / ID: 3 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 7.594898 KDa |
| Sequence | String: (DC)(DA)(DC)(DC)(DG)(DA)(DG)(DC)(DT)(DT) (DG)(DA)(DT)(DC)(DC)(DT)(DC)(DG)(DA)(DT) (DG)(DA)(DT)(DC)(DC) |
-Macromolecule #4: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 4 / Number of copies: 1 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blot for 3 seconds before plunging. | ||||||||||||
| Details | Protein sample was purified over a gel filtration column and mixed with DNA+AMP-PNP prior to grid preparation |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-40 / Number grids imaged: 1 / Number real images: 2351 / Average exposure time: 12.0 sec. / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Calibrated defocus max: 3.0 µm / Calibrated defocus min: 1.5 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 64000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |


