Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-10857: Cryo-EM structure of Tetrahymena thermophila mitochondrial ATP sy... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10857 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
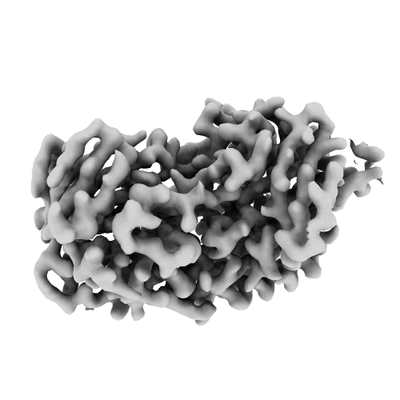
| Title | Cryo-EM structure of Tetrahymena thermophila mitochondrial ATP synthase - Fo-wing region | ||||||||||||
 Map data Map data | Local-resolution filtered full map of T. thermophila ATP synthase Fo-wing region | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | mitochondria / ATP synthase / oxidoreductase / NAD / MEMBRANE PROTEIN | ||||||||||||
| Function / homology | Sulphide quinone-reductase / : / sulfide:quinone oxidoreductase activity / FAD/NAD(P)-binding domain / Pyridine nucleotide-disulphide oxidoreductase / FAD binding / FAD/NAD(P)-binding domain superfamily / mitochondrion / Oxidoreductase, putative Function and homology information Function and homology information | ||||||||||||
| Biological species |   Tetrahymena thermophila (eukaryote) Tetrahymena thermophila (eukaryote) | ||||||||||||
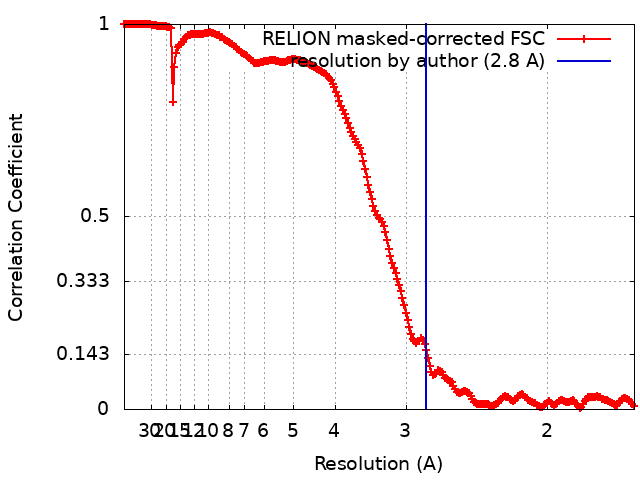
| Method | single particle reconstruction / cryo EM / Resolution: 2.8 Å | ||||||||||||
 Authors Authors | Kock Flygaard R / Muhleip A | ||||||||||||
| Funding support |  Sweden, 3 items Sweden, 3 items
| ||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2020 Title: Type III ATP synthase is a symmetry-deviated dimer that induces membrane curvature through tetramerization. Authors: Rasmus Kock Flygaard / Alexander Mühleip / Victor Tobiasson / Alexey Amunts / Abstract: Mitochondrial ATP synthases form functional homodimers to induce cristae curvature that is a universal property of mitochondria. To expand on the understanding of this fundamental phenomenon, we ...Mitochondrial ATP synthases form functional homodimers to induce cristae curvature that is a universal property of mitochondria. To expand on the understanding of this fundamental phenomenon, we characterized the unique type III mitochondrial ATP synthase in its dimeric and tetrameric form. The cryo-EM structure of a ciliate ATP synthase dimer reveals an unusual U-shaped assembly of 81 proteins, including a substoichiometrically bound ATPTT2, 40 lipids, and co-factors NAD and CoQ. A single copy of subunit ATPTT2 functions as a membrane anchor for the dimeric inhibitor IF. Type III specific linker proteins stably tie the ATP synthase monomers in parallel to each other. The intricate dimer architecture is scaffolded by an extended subunit-a that provides a template for both intra- and inter-dimer interactions. The latter results in the formation of tetramer assemblies, the membrane part of which we determined to 3.1 Å resolution. The structure of the type III ATP synthase tetramer and its associated lipids suggests that it is the intact unit propagating the membrane curvature. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10857.map.gz | 472.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10857-v30.xmlemd-10857.xml | 15.8 KB 15.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_10857_fsc.xml | 21.1 KB | Display | FSC data file |
| Images | 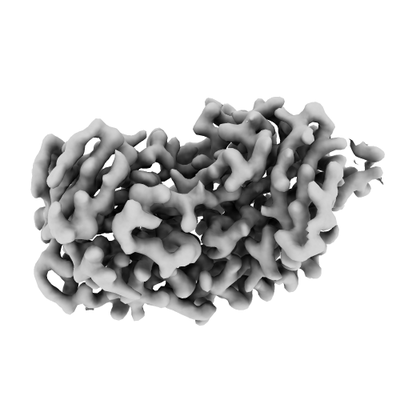 emd_10857.png emd_10857.png | 55.4 KB | ||
| Masks | emd_10857_msk_1.map | 824 MB | Mask map | |
| Filedesc metadata | emd-10857.cif.gz | 5.6 KB | ||
| Others | emd_10857_half_map_1.map.gzemd_10857_half_map_2.map.gz | 672.1 MB 669.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10857ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10857 http://ftp.pdbj.org/pub/emdb/structures/EMD-10857ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10857 | HTTPS FTP |
-Related structure data
| Related structure data |  6ynvMC  6ynwC  6ynxC  6ynyC  6ynzC  6yo0C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_10857.map.gz / Format: CCP4 / Size: 824 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Local-resolution filtered full map of T. thermophila ATP synthase Fo-wing region | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
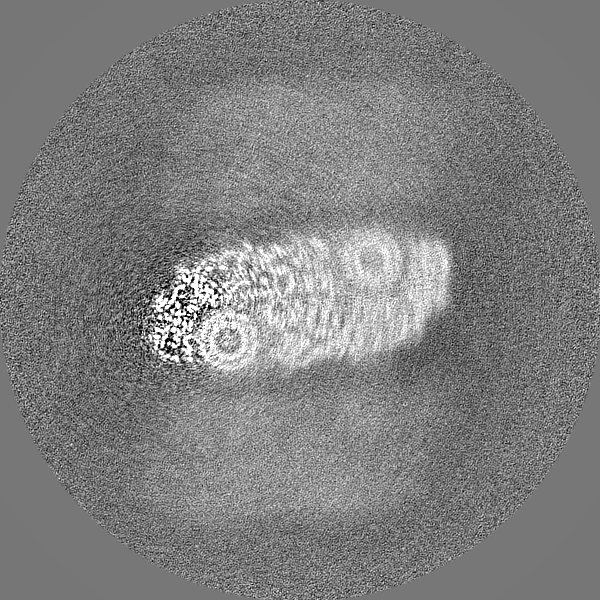
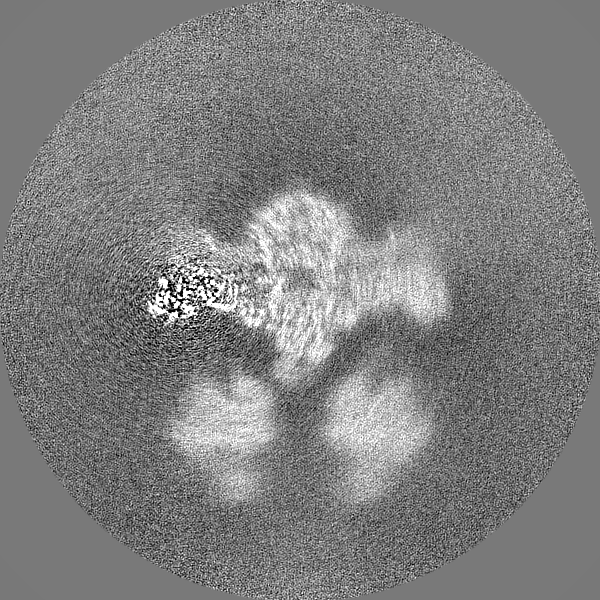
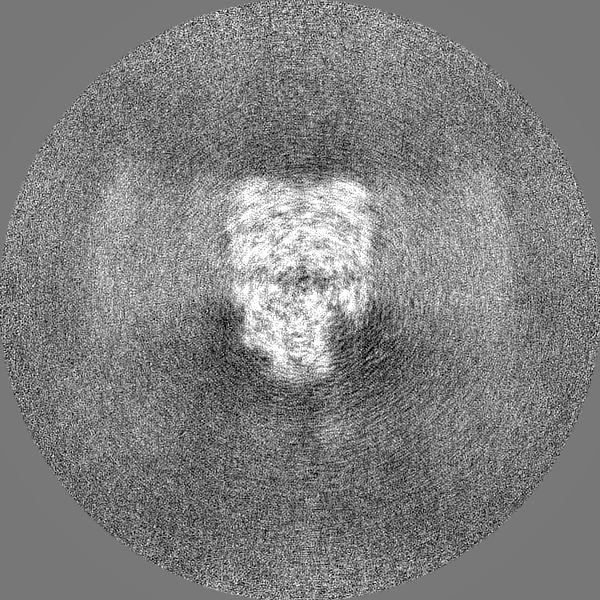
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.83 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
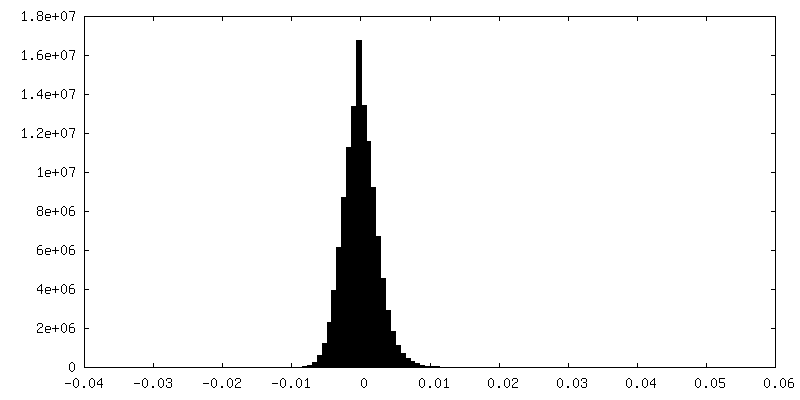
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_10857_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
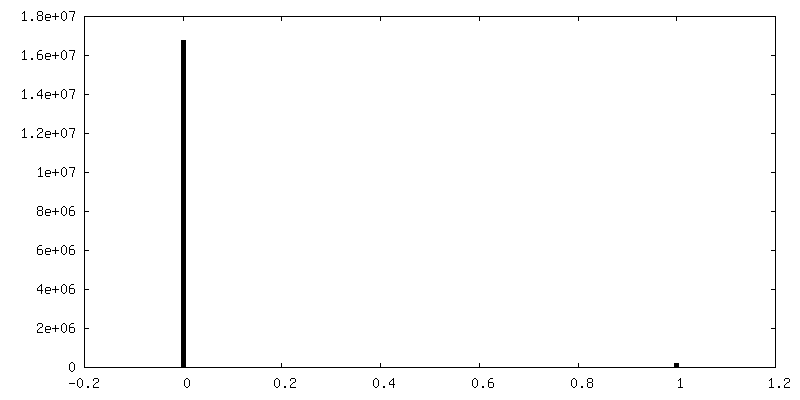
| Density Histograms |






-Half map: Half-map 1
| File | emd_10857_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 1 | ||||||||||||
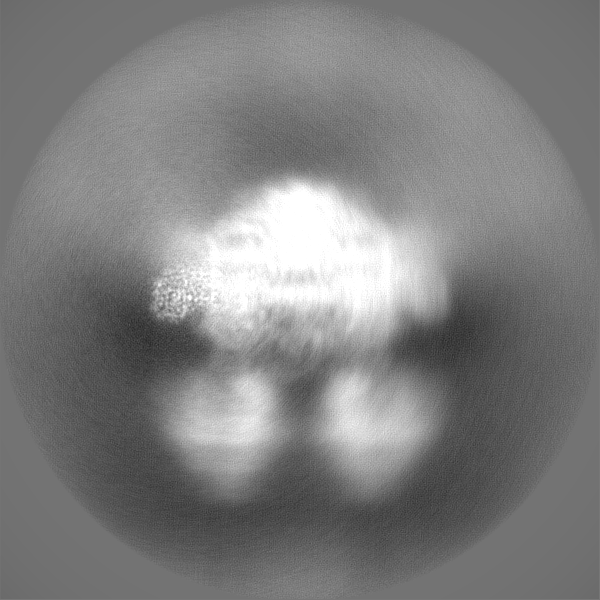
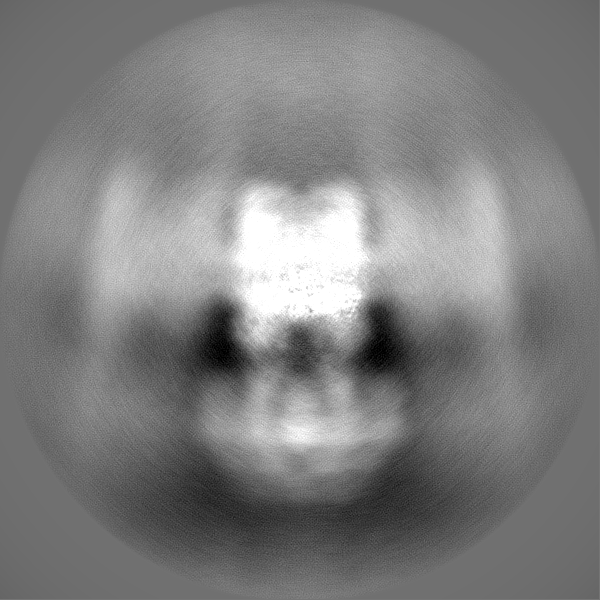
| Projections & Slices |
| ||||||||||||
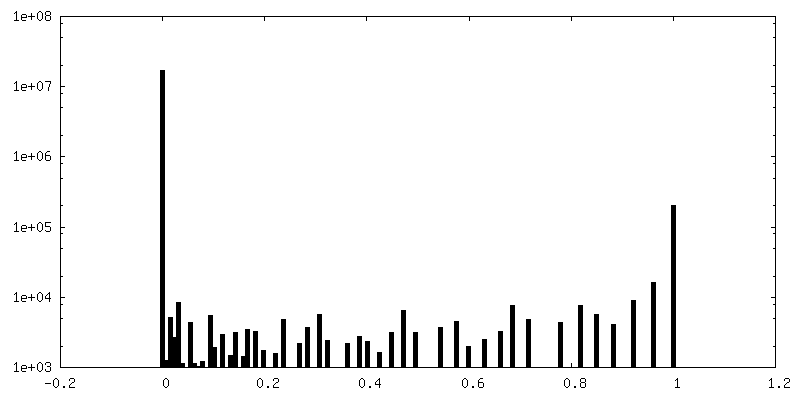
| Density Histograms |
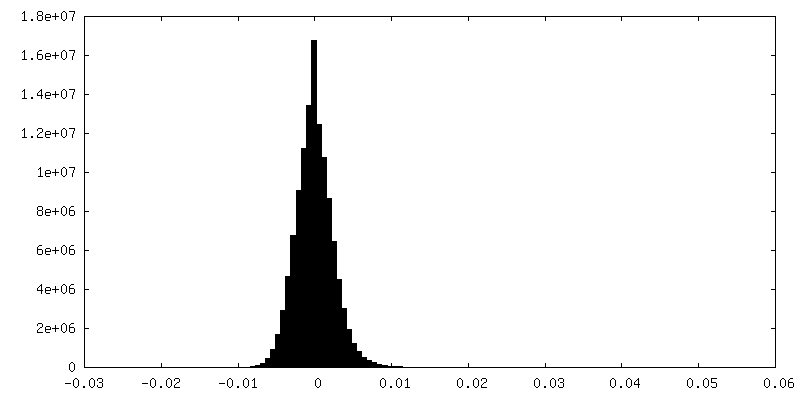
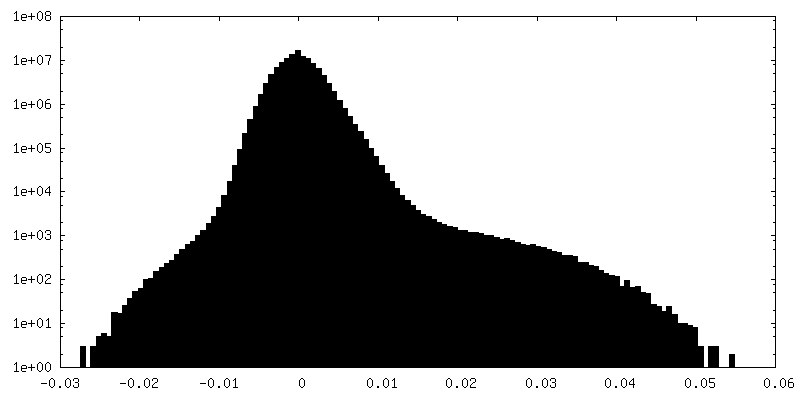
-Half map: Half-map 2
| File | emd_10857_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

- Sample components
Sample components
-Entire : Mitochondrial ATP synthase, Fo-wing region
| Entire | Name: Mitochondrial ATP synthase, Fo-wing region |
|---|---|
| Components |
|
-Supramolecule #1: Mitochondrial ATP synthase, Fo-wing region
| Supramolecule | Name: Mitochondrial ATP synthase, Fo-wing region / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
-Macromolecule #1: ATPTT1
| Macromolecule | Name: ATPTT1 / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
| Molecular weight | Theoretical: 54.917816 KDa |
| Sequence | String: MIHCLRNIRT VSALQSKISY NLGGGNKRKK TSGDLDNYDV LFVGANLGGI CSNHFDKDTH GKYKCFVSFD QPINQIYSVR IPYEQQRVR KSEYIHFSKK SINQFTPSEM LAVKEILPEQ NAVVLSSGRR IGYNQLVLAT GLKHDFSQIK GFYEALEHPE H PVYANRDP ...String: MIHCLRNIRT VSALQSKISY NLGGGNKRKK TSGDLDNYDV LFVGANLGGI CSNHFDKDTH GKYKCFVSFD QPINQIYSVR IPYEQQRVR KSEYIHFSKK SINQFTPSEM LAVKEILPEQ NAVVLSSGRR IGYNQLVLAT GLKHDFSQIK GFYEALEHPE H PVYANRDP ETWRSAQHKY SKYISNFKSG DGYFCIPEYP YAGEVECFNF FVSDEVWKWA QHHGALSPKH TFTIVNANEK FV HYCDSAD AFIKERLEKR GIRVEYNTKL LEVHQDGQKA TFINTKTGEK SVRDYNNLYS IVPSKRQEFL DKAGLTNGNG LLN VDHQTL QHKKYKNIFG LGDAADLPTT KTFWAGWYQI AVVRNNVKRN LQGQTLNAHY DGFSKVPLFT GHQTLTYVAH SYGG VGNWQ HLKHNNGGIL AWMRYRSWAK GMAKKFQDFY NGARLGPPYH KVLKSFPELP GSPESQQSSG ISKYFPTKTE NKAAH UniProtKB: Oxidoreductase, putative |
-Macromolecule #2: NICOTINAMIDE-ADENINE-DINUCLEOTIDE
| Macromolecule | Name: NICOTINAMIDE-ADENINE-DINUCLEOTIDE / type: ligand / ID: 2 / Number of copies: 1 / Formula: NAD |
|---|---|
| Molecular weight | Theoretical: 663.425 Da |
| Chemical component information |  ChemComp-NAD: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.75 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 300 |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: COUNTING / Average electron dose: 30.9 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal magnification: 165000 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |