 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-10816: Focused refinement cryo-EM structure of the yeast mitochondrial c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10816 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Focused refinement cryo-EM structure of the yeast mitochondrial complex I sub-stoichiometric sulfur transferase subunit | |||||||||
 Map data Map data | Focused refinement of sub-stoichiometric sulfur transferase | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | NADH:Ubiquinone Oxidoreductase / sulfur transferase / sub-stoichiometric / complex I / TRANSFERASE | |||||||||
| Function / homology |  Function and homology information Function and homology information3-mercaptopyruvate sulfurtransferase activity / tRNA wobble position uridine thiolation / thiosulfate-cyanide sulfurtransferase activity / mitochondrion Similarity search - Function | |||||||||
| Biological species |  Yarrowia lipolytica (yeast) Yarrowia lipolytica (yeast) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.5 Å | |||||||||
 Authors Authors | Hirst J / Grba D | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2020 Title: Mitochondrial complex I structure reveals ordered water molecules for catalysis and proton translocation. Authors: Daniel N Grba / Judy Hirst / Abstract: Mitochondrial complex I powers ATP synthesis by oxidative phosphorylation, exploiting the energy from ubiquinone reduction by NADH to drive protons across the energy-transducing inner membrane. ...Mitochondrial complex I powers ATP synthesis by oxidative phosphorylation, exploiting the energy from ubiquinone reduction by NADH to drive protons across the energy-transducing inner membrane. Recent cryo-EM analyses of mammalian and yeast complex I have revolutionized structural and mechanistic knowledge and defined structures in different functional states. Here, we describe a 2.7-Å-resolution structure of the 42-subunit complex I from the yeast Yarrowia lipolytica containing 275 structured water molecules. We identify a proton-relay pathway for ubiquinone reduction and water molecules that connect mechanistically crucial elements and constitute proton-translocation pathways through the membrane. By comparison with known structures, we deconvolute structural changes governing the mammalian 'deactive transition' (relevant to ischemia-reperfusion injury) and their effects on the ubiquinone-binding site and a connected cavity in ND1. Our structure thus provides important insights into catalysis by this enigmatic respiratory machine. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10816.map.gz | 323.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10816-v30.xmlemd-10816.xml | 19.2 KB 19.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_10816_fsc.xml | 15.9 KB | Display | FSC data file |
| Images |  emd_10816.png emd_10816.png | 78.6 KB | ||
| Masks | emd_10816_msk_1.map | 347.6 MB | Mask map | |
| Filedesc metadata | emd-10816.cif.gz | 6.4 KB | ||
| Others | emd_10816_half_map_1.map.gzemd_10816_half_map_2.map.gz | 276.5 MB 276.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10816ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10816 http://ftp.pdbj.org/pub/emdb/structures/EMD-10816ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10816 | HTTPS FTP |
-Related structure data
| Related structure data |  6yj5MC  6skkC  6skmC  6sknC  6slqC  6sluC  6smuC  6y9vC  6y9wC  6y9xC  6y9yC  6y9zC  6zdjC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_10816.map.gz / Format: CCP4 / Size: 347.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Focused refinement of sub-stoichiometric sulfur transferase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.055 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
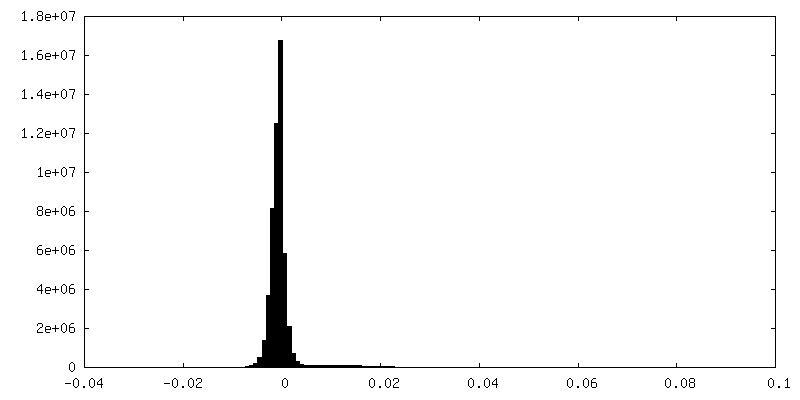
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_10816_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
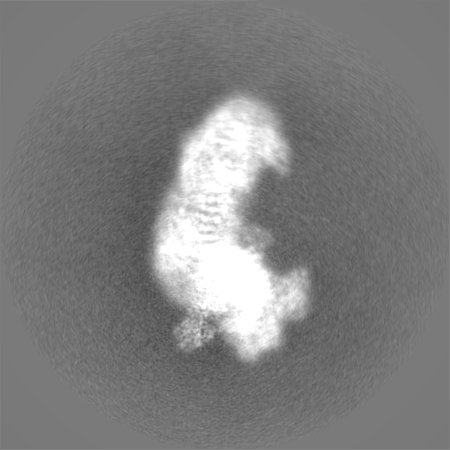
| Projections & Slices |
| ||||||||||||
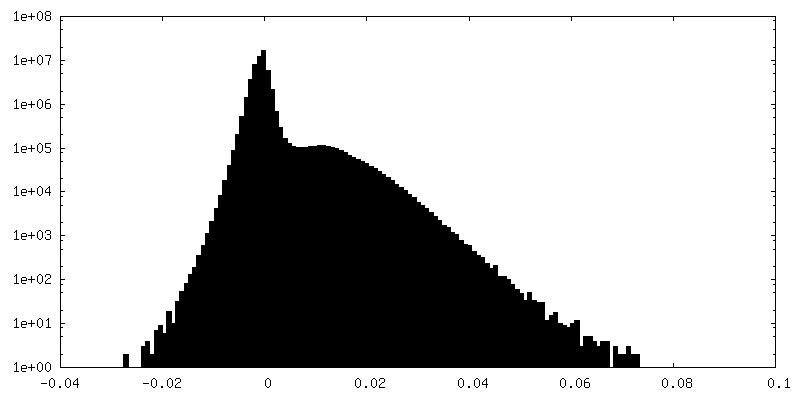
| Density Histograms |






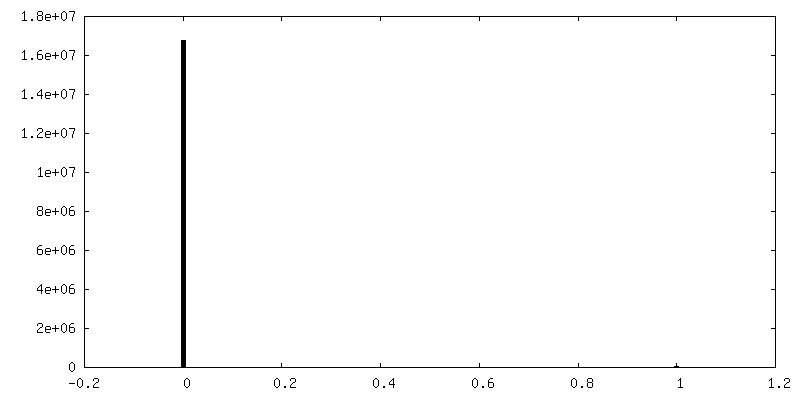

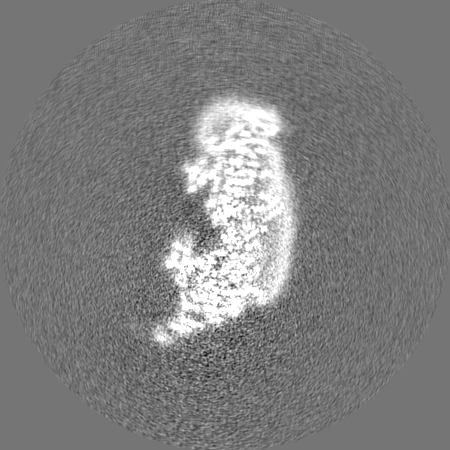
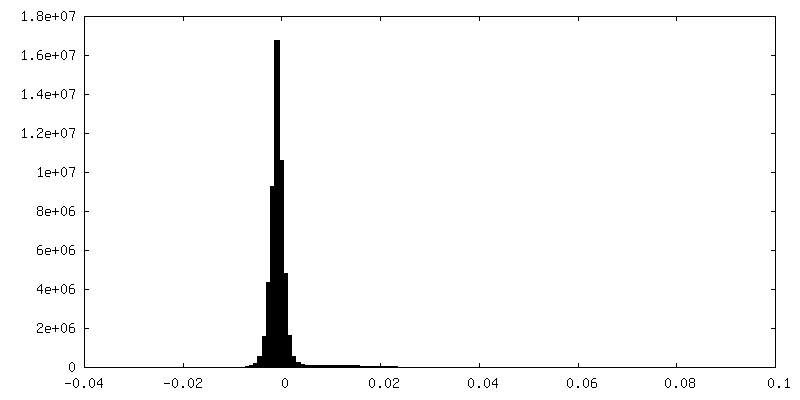
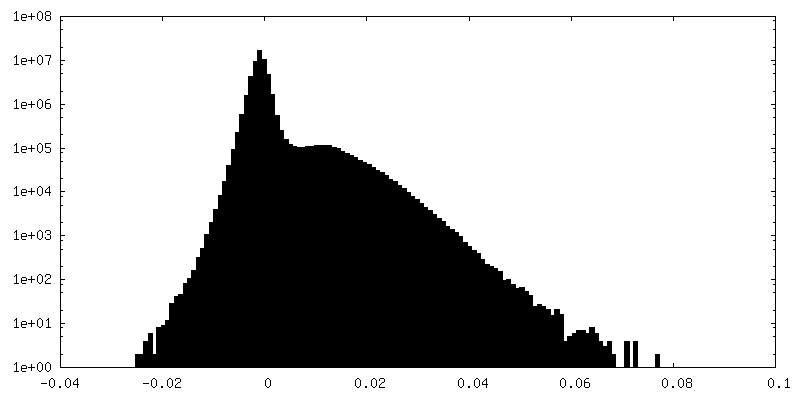
-Half map: Half map 2
| File | emd_10816_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
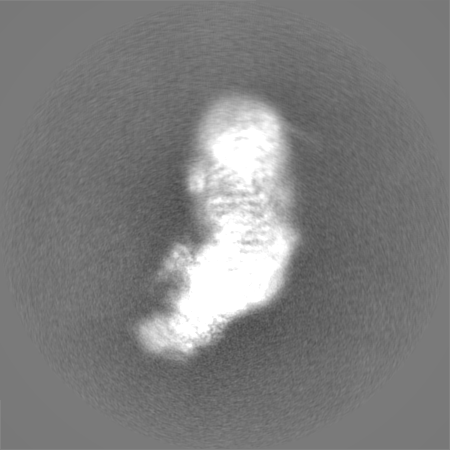
| Projections & Slices |
| ||||||||||||
| Density Histograms |


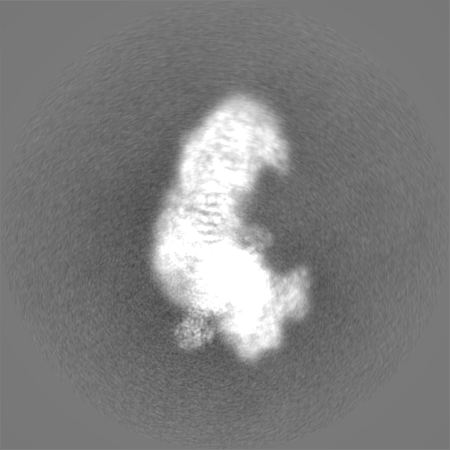
-Half map: Half map 1
| File | emd_10816_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
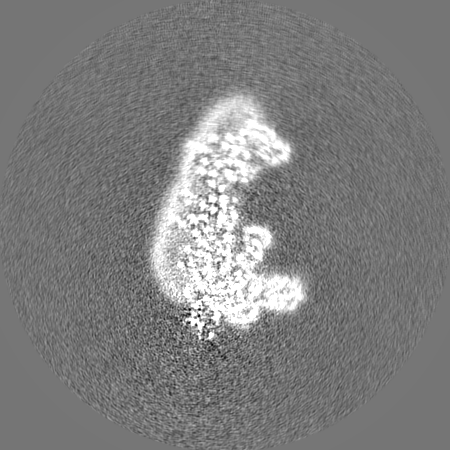
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : ST1 (sulfur transferase)
| Entire | Name: ST1 (sulfur transferase) |
|---|---|
| Components |
|
-Supramolecule #1: ST1 (sulfur transferase)
| Supramolecule | Name: ST1 (sulfur transferase) / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Yarrowia lipolytica (yeast) |
-Macromolecule #1: Rhodanese-like domain-containing protein
| Macromolecule | Name: Rhodanese-like domain-containing protein / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Yarrowia lipolytica (yeast) |
| Molecular weight | Theoretical: 34.661117 KDa |
| Recombinant expression | Organism: Yarrowia lipolytica (yeast) |
| Sequence | String: MSKLISPAEL AKRLSSKETK IFDATWYLPT PANVGKNAYD NYMKKRIPGA LYFDIDAVNT PSKFPHMLPS PQTFENELTK LGVSSDSPI VVYDTQGVFS GPRLVWTFKV FGHDNVQFLN GFEAYTQLPG IPSRPDAYTW GIWDTQVPGK IDPADPPYKV T KARPELVK ...String: MSKLISPAEL AKRLSSKETK IFDATWYLPT PANVGKNAYD NYMKKRIPGA LYFDIDAVNT PSKFPHMLPS PQTFENELTK LGVSSDSPI VVYDTQGVFS GPRLVWTFKV FGHDNVQFLN GFEAYTQLPG IPSRPDAYTW GIWDTQVPGK IDPADPPYKV T KARPELVK SFEDVLAIVE KHNGDGAKIR NEVTFIDARP NGRFTGKDAE PRAELSSGHV PGAYSIAFPE VVENGKFKSP EE LKALFAS KGIDGSKPII SMCGSGVTAC VIDLALEIAG IGSRDTNAVY DGSWTEWAQR APTKYIVKEE NLNEANRA UniProtKB: Rhodanese-like domain-containing protein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.45 |
|---|---|
| Grid | Model: UltrAuFoil / Material: GOLD / Support film - Material: GOLD / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 90 sec. / Details: Gold grids saturated with PEG thiol reagent |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 2540 / Average electron dose: 44.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: -2.7 µm / Calibrated defocus min: -1.5 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 130000 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | SWISS-MODEL used to generate initial model using PDB 1RHD as a template |
| Output model | PDB-6yj5: |

