 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0568: Cryo-EM 3D map of human ATP-citrate lyase N-terminal segment in c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0568 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM 3D map of human ATP-citrate lyase N-terminal segment in complex with inhibitor NDI-091143 | |||||||||
 Map data Map data | em-volume_P1 | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
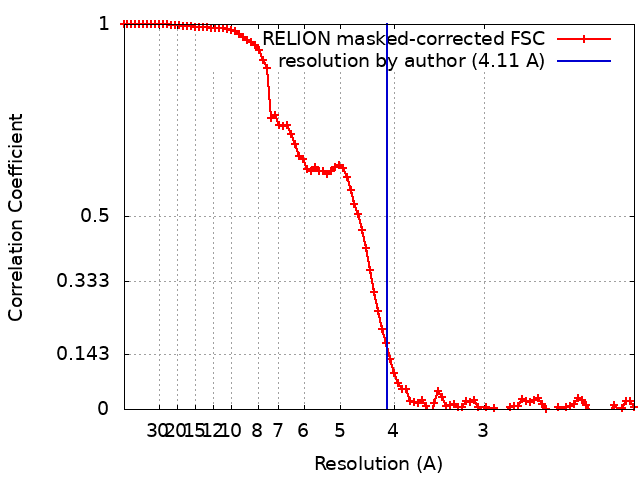
| Method | single particle reconstruction / cryo EM / Resolution: 4.11 Å | |||||||||
 Authors Authors | Wei J / Tong L | |||||||||
 Citation Citation | Journal: Nature / Year: 2019 Title: An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Authors: Jia Wei / Silvana Leit / Jun Kuai / Eric Therrien / Salma Rafi / H James Harwood / Byron DeLaBarre / Liang Tong /  Abstract: ATP-citrate lyase (ACLY) is a central metabolic enzyme and catalyses the ATP-dependent conversion of citrate and coenzyme A (CoA) to oxaloacetate and acetyl-CoA. The acetyl-CoA product is crucial for ...ATP-citrate lyase (ACLY) is a central metabolic enzyme and catalyses the ATP-dependent conversion of citrate and coenzyme A (CoA) to oxaloacetate and acetyl-CoA. The acetyl-CoA product is crucial for the metabolism of fatty acids, the biosynthesis of cholesterol, and the acetylation and prenylation of proteins. There has been considerable interest in ACLY as a target for anti-cancer drugs, because many cancer cells depend on its activity for proliferation. ACLY is also a target against dyslipidaemia and hepatic steatosis, with a compound currently in phase 3 clinical trials. Many inhibitors of ACLY have been reported, but most of them have weak activity. Here we report the development of a series of low nanomolar, small-molecule inhibitors of human ACLY. We have also determined the structure of the full-length human ACLY homo-tetramer in complex with one of these inhibitors (NDI-091143) by cryo-electron microscopy, which reveals an unexpected mechanism of inhibition. The compound is located in an allosteric, mostly hydrophobic cavity next to the citrate-binding site, and requires extensive conformational changes in the enzyme that indirectly disrupt citrate binding. The observed binding mode is supported by and explains the structure-activity relationships of these compounds. This allosteric site greatly enhances the 'druggability' of ACLY and represents an attractive target for the development of new ACLY inhibitors. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0568.map.gz | 4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0568-v30.xmlemd-0568.xml | 15.8 KB 15.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_0568_fsc.xml | 9.2 KB | Display | FSC data file |
| Images |  emd_0568.png emd_0568.png | 35.9 KB | ||
| Masks | emd_0568_msk_1.map | 64 MB | Mask map | |
| Others | emd_0568_half_map_1.map.gzemd_0568_half_map_2.map.gz | 48.4 MB 48.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0568ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0568 http://ftp.pdbj.org/pub/emdb/structures/EMD-0568ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0568 | HTTPS FTP |
-Related structure data










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0568.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | em-volume_P1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.0605 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
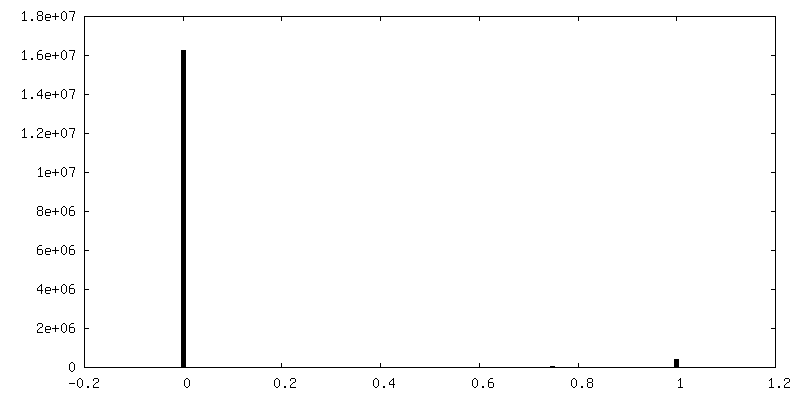
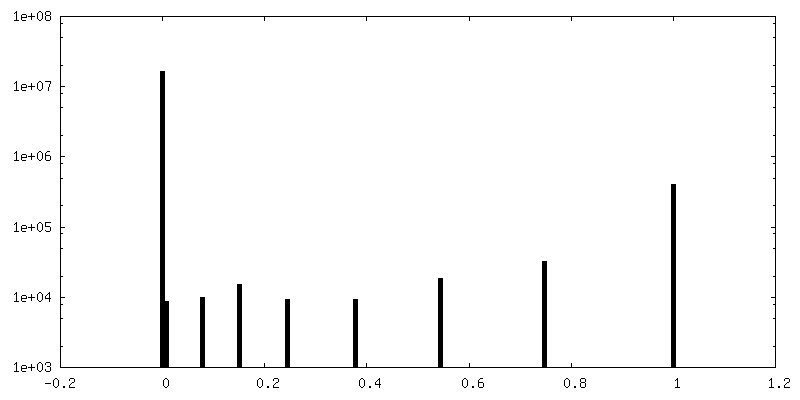
-Mask #1
| File | emd_0568_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
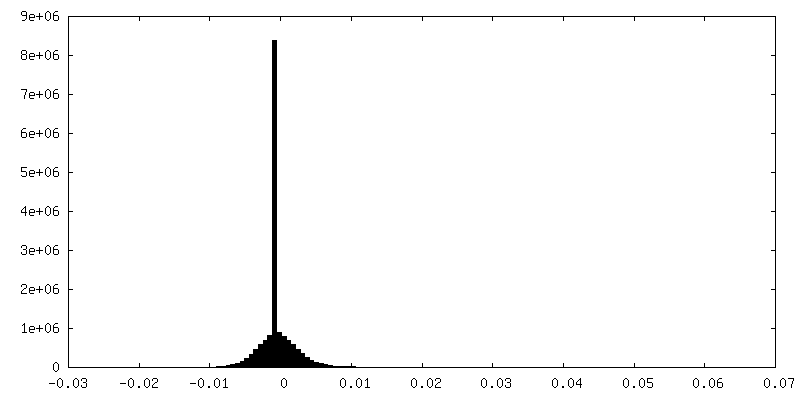
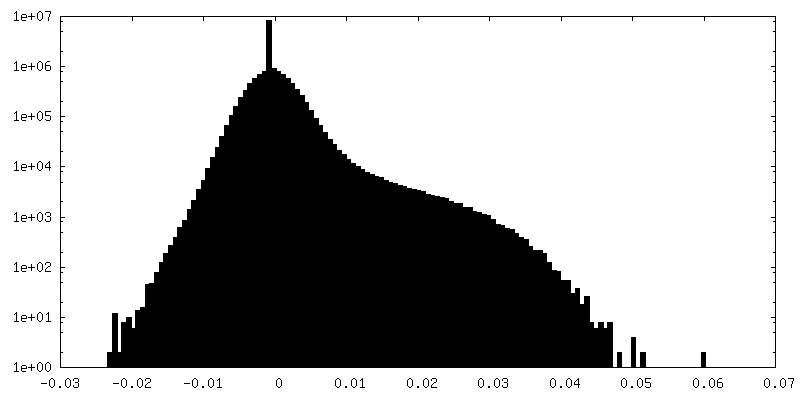
| Density Histograms |






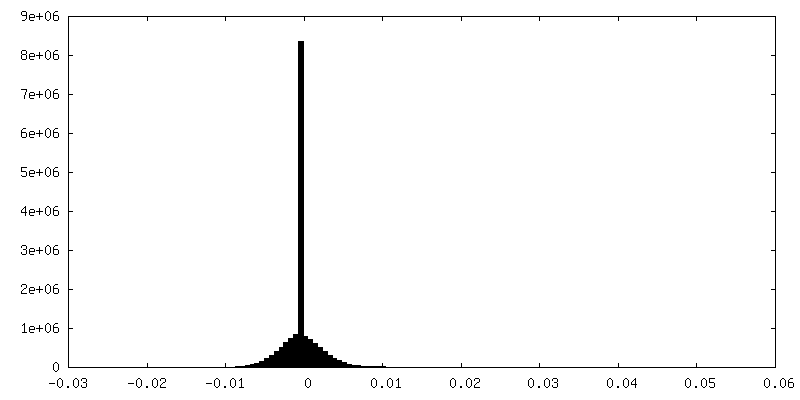
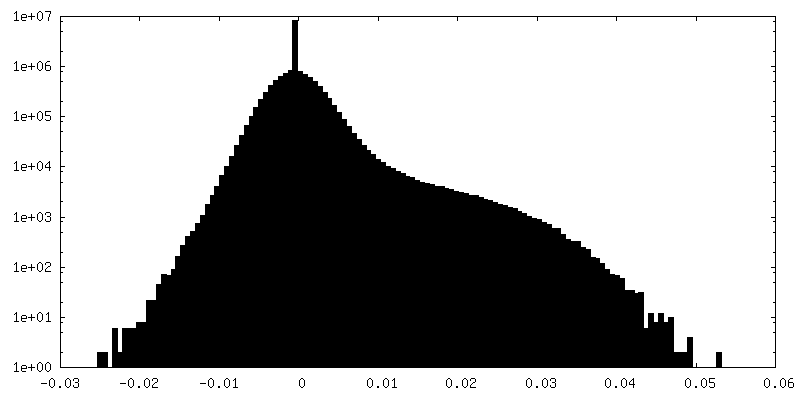
-Half map: em-half-volume P1
| File | emd_0568_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | em-half-volume_P1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: em-half-volume P2
| File | emd_0568_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | em-half-volume_P2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : protein-inhibitor complex
| Entire | Name: protein-inhibitor complex |
|---|---|
| Components |
|
-Supramolecule #1: protein-inhibitor complex
| Supramolecule | Name: protein-inhibitor complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism:  |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4.7 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295 K / Instrument: FEI VITROBOT MARK II |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number real images: 2803 / Average exposure time: 10.0 sec. / Average electron dose: 71.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: -0.002 µm / Nominal defocus min: -0.0008 µm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: AB INITIO MODEL |
|---|
