 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 8g9b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Human IMPDH2 mutant - L245P, treated with GTP, ATP, IMP, and NAD+; compressed filament segment reconstruction | |||||||||
 要素 要素 | Inosine-5'-monophosphate dehydrogenase 2 | |||||||||
 キーワード キーワード | OXIDOREDUCTASE / Filament / Dehydrogenase / CBS domain / Bateman domain / purine biosynthesis | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / lymphocyte proliferation / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / peroxisomal membrane / Azathioprine ADME / GTP biosynthetic process / cellular response to interleukin-4 ...'de novo' XMP biosynthetic process / Purine ribonucleoside monophosphate biosynthesis / lymphocyte proliferation / IMP dehydrogenase / IMP dehydrogenase activity / GMP biosynthetic process / peroxisomal membrane / Azathioprine ADME / GTP biosynthetic process / cellular response to interleukin-4 / circadian rhythm / secretory granule lumen / ficolin-1-rich granule lumen / Potential therapeutics for SARS / nucleotide binding / Neutrophil degranulation / DNA binding / RNA binding / extracellular exosome / extracellular region / metal ion binding / nucleus / membrane / cytosol / cytoplasm 類似検索 - 分子機能 | |||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||
| 手法 | 電子顕微鏡法 / 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3 Å | |||||||||
 データ登録者 データ登録者 | O'Neill, A.G. / Kollman, J.M. | |||||||||
| 資金援助 |  米国, 2件 米国, 2件
| |||||||||
 引用 引用 | ジャーナル: J Biol Chem / 年: 2023 タイトル: Neurodevelopmental disorder mutations in the purine biosynthetic enzyme IMPDH2 disrupt its allosteric regulation. 著者: Audrey G O'Neill / Anika L Burrell / Michael Zech / Orly Elpeleg / Tamar Harel / Simon Edvardson / Hagar Mor-Shaked / Alyssa L Rippert / Tomoki Nomakuchi / Kosuke Izumi / Justin M Kollman /   要旨: Inosine 5' monophosphate dehydrogenase (IMPDH) is a critical regulatory enzyme in purine nucleotide biosynthesis that is inhibited by the downstream product GTP. Multiple point mutations in the human ...Inosine 5' monophosphate dehydrogenase (IMPDH) is a critical regulatory enzyme in purine nucleotide biosynthesis that is inhibited by the downstream product GTP. Multiple point mutations in the human isoform IMPDH2 have recently been associated with dystonia and other neurodevelopmental disorders, but the effect of the mutations on enzyme function has not been described. Here, we report the identification of two additional missense variants in IMPDH2 from affected individuals and show that all of the disease-associated mutations disrupt GTP regulation. Cryo-EM structures of one IMPDH2 mutant suggest this regulatory defect arises from a shift in the conformational equilibrium toward a more active state. This structural and functional analysis provides insight into IMPDH2-associated disease mechanisms that point to potential therapeutic approaches and raises new questions about fundamental aspects of IMPDH regulation. #1: ジャーナル: bioRxiv / 年: 2023 タイトル: Point mutations in IMPDH2 which cause early-onset neurodevelopmental disorders disrupt enzyme regulation and filament structure. 著者: Audrey G O'Neill / Anika L Burrell / Michael Zech / Orly Elpeleg / Tamar Harel / Simon Edvardson / Hagar Mor Shaked / Alyssa L Rippert / Tomoki Nomakuchi / Kosuke Izumi / Justin M Kollman / 要旨: Inosine 5' monophosphate dehydrogenase (IMPDH) is a critical regulatory enzyme in purine nucleotide biosynthesis that is inhibited by the downstream product GTP. Multiple point mutations in the human ...Inosine 5' monophosphate dehydrogenase (IMPDH) is a critical regulatory enzyme in purine nucleotide biosynthesis that is inhibited by the downstream product GTP. Multiple point mutations in the human isoform IMPDH2 have recently been associated with dystonia and other neurodevelopmental disorders, but the effect of the mutations on enzyme function has not been described. Here, we report identification of two additional affected individuals with missense variants in and show that all of the disease-associated mutations disrupt GTP regulation. Cryo-EM structures of one IMPDH2 mutant suggest this regulatory defect arises from a shift in the conformational equilibrium toward a more active state. This structural and functional analysis provides insight into IMPDH2-associated disease mechanisms that point to potential therapeutic approaches and raises new questions about fundamental aspects of IMPDH regulation. #2: ジャーナル: Elife / 年: 2018タイトル: New tools for automated high-resolution cryo-EM structure determination in RELION-3. 著者: Jasenko Zivanov / Takanori Nakane / Björn O Forsberg / Dari Kimanius / Wim Jh Hagen / Erik Lindahl / Sjors Hw Scheres /   要旨: Here, we describe the third major release of RELION. CPU-based vector acceleration has been added in addition to GPU support, which provides flexibility in use of resources and avoids memory ...Here, we describe the third major release of RELION. CPU-based vector acceleration has been added in addition to GPU support, which provides flexibility in use of resources and avoids memory limitations. Reference-free autopicking with Laplacian-of-Gaussian filtering and execution of jobs from python allows non-interactive processing during acquisition, including 2D-classification, model generation and 3D-classification. Per-particle refinement of CTF parameters and correction of estimated beam tilt provides higher resolution reconstructions when particles are at different heights in the ice, and/or coma-free alignment has not been optimal. Ewald sphere curvature correction improves resolution for large particles. We illustrate these developments with publicly available data sets: together with a Bayesian approach to beam-induced motion correction it leads to resolution improvements of 0.2-0.7 Å compared to previous RELION versions. #3: ジャーナル: J Struct Biol / 年: 2012 タイトル: RELION: implementation of a Bayesian approach to cryo-EM structure determination. 著者: Sjors H W Scheres / 要旨: RELION, for REgularized LIkelihood OptimizatioN, is an open-source computer program for the refinement of macromolecular structures by single-particle analysis of electron cryo-microscopy (cryo-EM) ...RELION, for REgularized LIkelihood OptimizatioN, is an open-source computer program for the refinement of macromolecular structures by single-particle analysis of electron cryo-microscopy (cryo-EM) data. Whereas alternative approaches often rely on user expertise for the tuning of parameters, RELION uses a Bayesian approach to infer parameters of a statistical model from the data. This paper describes developments that reduce the computational costs of the underlying maximum a posteriori (MAP) algorithm, as well as statistical considerations that yield new insights into the accuracy with which the relative orientations of individual particles may be determined. A so-called gold-standard Fourier shell correlation (FSC) procedure to prevent overfitting is also described. The resulting implementation yields high-quality reconstructions and reliable resolution estimates with minimal user intervention and at acceptable computational costs. #4: ジャーナル: Nat Methods / 年: 2017 タイトル: MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. 著者: Shawn Q Zheng / Eugene Palovcak / Jean-Paul Armache / Kliment A Verba / Yifan Cheng / David A Agard / #5: ジャーナル: J Struct Biol / 年: 2000 タイトル: Leginon: an automated system for acquisition of images from vitreous ice specimens. 著者: B Carragher / N Kisseberth / D Kriegman / R A Milligan / C S Potter / J Pulokas / A Reilein / 要旨: We have developed a system to automatically acquire cryo-electron micrographs. The system is designed to emulate all of the decisions and actions of a highly trained microscopist in collecting data ...We have developed a system to automatically acquire cryo-electron micrographs. The system is designed to emulate all of the decisions and actions of a highly trained microscopist in collecting data from a vitreous ice specimen. These include identifying suitable areas of vitreous ice at low magnification, determining the presence and location of specimen on the grid, automatically adjusting imaging parameters (focus, astigmatism) under low-dose conditions, and acquiring images at high magnification to either film or a digital camera. This system is responsible for every aspect of image acquisition and can run unattended, other than requiring periodic refilling of the cryogens, for over 24 h. The system has been tested out on a variety of specimens that represent typical challenges in the field of cryo-electron microscopy. The results show that the overall performance of the system is equivalent to that of an experienced microscopist. #6: ジャーナル: J Struct Biol / 年: 2015 タイトル: CTFFIND4: Fast and accurate defocus estimation from electron micrographs. 著者: Alexis Rohou / Nikolaus Grigorieff / 要旨: CTFFIND is a widely-used program for the estimation of objective lens defocus parameters from transmission electron micrographs. Defocus parameters are estimated by fitting a model of the ...CTFFIND is a widely-used program for the estimation of objective lens defocus parameters from transmission electron micrographs. Defocus parameters are estimated by fitting a model of the microscope's contrast transfer function (CTF) to an image's amplitude spectrum. Here we describe modifications to the algorithm which make it significantly faster and more suitable for use with images collected using modern technologies such as dose fractionation and phase plates. We show that this new version preserves the accuracy of the original algorithm while allowing for higher throughput. We also describe a measure of the quality of the fit as a function of spatial frequency and suggest this can be used to define the highest resolution at which CTF oscillations were successfully modeled. #7: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2019 タイトル: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. 著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / ...著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /  要旨: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. #8: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2018 タイトル: Real-space refinement in PHENIX for cryo-EM and crystallography. 著者: Pavel V Afonine / Billy K Poon / Randy J Read / Oleg V Sobolev / Thomas C Terwilliger / Alexandre Urzhumtsev / Paul D Adams / 要旨: This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast ...This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps. #9: ジャーナル: Nat Methods / 年: 2020 タイトル: Improvement of cryo-EM maps by density modification. 著者: Thomas C Terwilliger / Steven J Ludtke / Randy J Read / Paul D Adams / Pavel V Afonine / 要旨: A density-modification procedure for improving maps from single-particle electron cryogenic microscopy (cryo-EM) is presented. The theoretical basis of the method is identical to that of maximum- ...A density-modification procedure for improving maps from single-particle electron cryogenic microscopy (cryo-EM) is presented. The theoretical basis of the method is identical to that of maximum-likelihood density modification, previously used to improve maps from macromolecular X-ray crystallography. Key differences from applications in crystallography are that the errors in Fourier coefficients are largely in the phases in crystallography but in both phases and amplitudes in cryo-EM, and that half-maps with independent errors are available in cryo-EM. These differences lead to a distinct approach for combination of information from starting maps with information obtained in the density-modification process. The density-modification procedure was applied to a set of 104 datasets and improved map-model correlation and increased the visibility of details in many of the maps. The procedure requires two unmasked half-maps and a sequence file or other source of information on the volume of the macromolecule that has been imaged. #10: ジャーナル: Acta Crystallogr D Biol Crystallogr / 年: 2004 タイトル: Coot: model-building tools for molecular graphics. 著者: Paul Emsley / Kevin Cowtan / 要旨: CCP4mg is a project that aims to provide a general-purpose tool for structural biologists, providing tools for X-ray structure solution, structure comparison and analysis, and publication-quality ...CCP4mg is a project that aims to provide a general-purpose tool for structural biologists, providing tools for X-ray structure solution, structure comparison and analysis, and publication-quality graphics. The map-fitting tools are available as a stand-alone package, distributed as 'Coot'. #11: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2018タイトル: ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. 著者: Tristan Ian Croll / 要旨: This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density ...This paper introduces ISOLDE, a new software package designed to provide an intuitive environment for high-fidelity interactive remodelling/refinement of macromolecular models into electron-density maps. ISOLDE combines interactive molecular-dynamics flexible fitting with modern molecular-graphics visualization and established structural biology libraries to provide an immersive interface wherein the model constantly acts to maintain physically realistic conformations as the user interacts with it by directly tugging atoms with a mouse or haptic interface or applying/removing restraints. In addition, common validation tasks are accelerated and visualized in real time. Using the recently described 3.8 Å resolution cryo-EM structure of the eukaryotic minichromosome maintenance (MCM) helicase complex as a case study, it is demonstrated how ISOLDE can be used alongside other modern refinement tools to avoid common pitfalls of low-resolution modelling and improve the quality of the final model. A detailed analysis of changes between the initial and final model provides a somewhat sobering insight into the dangers of relying on a small number of validation metrics to judge the quality of a low-resolution model. #12: ジャーナル: J Comput Chem / 年: 2004 タイトル: UCSF Chimera--a visualization system for exploratory research and analysis. 著者: Eric F Pettersen / Thomas D Goddard / Conrad C Huang / Gregory S Couch / Daniel M Greenblatt / Elaine C Meng / Thomas E Ferrin / 要旨: The design, implementation, and capabilities of an extensible visualization system, UCSF Chimera, are discussed. Chimera is segmented into a core that provides basic services and visualization, and ...The design, implementation, and capabilities of an extensible visualization system, UCSF Chimera, are discussed. Chimera is segmented into a core that provides basic services and visualization, and extensions that provide most higher level functionality. This architecture ensures that the extension mechanism satisfies the demands of outside developers who wish to incorporate new features. Two unusual extensions are presented: Multiscale, which adds the ability to visualize large-scale molecular assemblies such as viral coats, and Collaboratory, which allows researchers to share a Chimera session interactively despite being at separate locales. Other extensions include Multalign Viewer, for showing multiple sequence alignments and associated structures; ViewDock, for screening docked ligand orientations; Movie, for replaying molecular dynamics trajectories; and Volume Viewer, for display and analysis of volumetric data. A discussion of the usage of Chimera in real-world situations is given, along with anticipated future directions. Chimera includes full user documentation, is free to academic and nonprofit users, and is available for Microsoft Windows, Linux, Apple Mac OS X, SGI IRIX, and HP Tru64 Unix from http://www.cgl.ucsf.edu/chimera/. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 8g9b.cif.gz | 751.1 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb8g9b.ent.gz | 630 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 8g9b.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 8g9b_validation.pdf.gz | 3.6 MB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 8g9b_full_validation.pdf.gz | 3.6 MB | 表示 | |
| XML形式データ | 8g9b_validation.xml.gz | 126.2 KB | 表示 | |
| CIF形式データ | 8g9b_validation.cif.gz | 175.6 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/g9/8g9bftp://data.pdbj.org/pub/pdb/validation_reports/g9/8g9b | HTTPS FTP |
-関連構造データ








-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
|
|---|---|
| 1 |
|
-要素
| #1: タンパク質 | 分子量: 56464.453 Da / 分子数: 8 / 変異: L245P / 由来タイプ: 組換発現 / 由来: (組換発現) Homo sapiens (ヒト) / 遺伝子: IMPDH2, IMPD2 / 発現宿主:  #2: 化合物 | ChemComp-GTP /   分子量: 523.180 Da / 分子数: 16 / 由来タイプ: 合成 / 式: C10H16N5O14P3 / タイプ: SUBJECT OF INVESTIGATION / コメント: GTP, エネルギー貯蔵分子*YM 分子量: 523.180 Da / 分子数: 16 / 由来タイプ: 合成 / 式: C10H16N5O14P3 / タイプ: SUBJECT OF INVESTIGATION / コメント: GTP, エネルギー貯蔵分子*YM#3: 化合物 | ChemComp-ATP /   分子量: 507.181 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C10H16N5O13P3 / タイプ: SUBJECT OF INVESTIGATION / コメント: ATP, エネルギー貯蔵分子*YM 分子量: 507.181 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C10H16N5O13P3 / タイプ: SUBJECT OF INVESTIGATION / コメント: ATP, エネルギー貯蔵分子*YM#4: 化合物 | ChemComp-IMP / 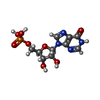  分子量: 348.206 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C10H13N4O8P / タイプ: SUBJECT OF INVESTIGATION 分子量: 348.206 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C10H13N4O8P / タイプ: SUBJECT OF INVESTIGATION#5: 化合物 | ChemComp-NAD /   分子量: 663.425 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C21H27N7O14P2 / タイプ: SUBJECT OF INVESTIGATION / コメント: NAD*YM 分子量: 663.425 Da / 分子数: 8 / 由来タイプ: 合成 / 式: C21H27N7O14P2 / タイプ: SUBJECT OF INVESTIGATION / コメント: NAD*YM研究の焦点であるリガンドがあるか | Y | Has protein modification | Y | |
|---|
-実験情報
-実験
| 実験 | 手法: 電子顕微鏡法 |
|---|---|
| EM実験 | 試料の集合状態: FILAMENT / 3次元再構成法: 単粒子再構成法 |
- 試料調製
試料調製
| 構成要素 | 名称: Filament of Inosine-5'-monophosphate dehydrogenase 2 - L245P mutant, bound to GTP, ATP, IMP, and NAD+ タイプ: COMPLEX 詳細: 5 uM enzyme was mixed with 20 mM GTP, 1 mM ATP, 1 mM MgCl2, 3 mM IMP, and 5 mM NAD+. Entity ID: #1 / 由来: RECOMBINANT |
|---|---|
| 分子量 | 実験値: NO |
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
| 由来(組換発現) | 生物種: |
| 緩衝液 | pH: 7 |
| 試料 | 包埋: NO / シャドウイング: NO / 染色: NO / 凍結: YES |
| 急速凍結 | 凍結剤: ETHANE |
- 電子顕微鏡撮影
電子顕微鏡撮影
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
|---|---|
| 顕微鏡 | モデル: FEI TITAN KRIOS |
| 電子銃 | 電子線源:  FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM |
| 電子レンズ | モード: BRIGHT FIELD / 倍率(公称値): 105000 X / 最大 デフォーカス(公称値): 280 nm / 最小 デフォーカス(公称値): 1740 nm / Cs: 2.7 mm |
| 試料ホルダ | 凍結剤: NITROGEN |
| 撮影 | 平均露光時間: 3 sec. / 電子線照射量: 60 e/Å2 / フィルム・検出器のモデル: GATAN K3 (6k x 4k) / 実像数: 2967 |
- 解析
解析
| EMソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF補正 | タイプ: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| 粒子像の選択 | 選択した粒子像数: 1421990 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 点対称性: D4 (2回x4回 2面回転対称) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3次元再構成 | 解像度: 3 Å / 解像度の算出法: OTHER / 粒子像の数: 26540 / 詳細: FSCref 0.5 cut-off from PHENIX density modification / 対称性のタイプ: POINT | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子モデル構築 | プロトコル: FLEXIBLE FIT / 空間: REAL | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
|
