 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6g0l: Structure of two molecules of the chromatin remodelling enzyme Ch... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6g0l | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of two molecules of the chromatin remodelling enzyme Chd1 bound to a nucleosome | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MOTOR PROTEIN / Chromatin remodellers | ||||||
| Function / homology |  Function and homology information Function and homology informationnucleolar chromatin / regulation of transcriptional start site selection at RNA polymerase II promoter / negative regulation of DNA-templated DNA replication / regulation of chromatin organization / rDNA binding / SLIK (SAGA-like) complex / DNA double-strand break processing / nucleosome organization / SAGA complex / sister chromatid cohesion ...nucleolar chromatin / regulation of transcriptional start site selection at RNA polymerase II promoter / negative regulation of DNA-templated DNA replication / regulation of chromatin organization / rDNA binding / SLIK (SAGA-like) complex / DNA double-strand break processing / nucleosome organization / SAGA complex / sister chromatid cohesion / ATP-dependent chromatin remodeler activity / termination of RNA polymerase II transcription / termination of RNA polymerase I transcription / ATP-dependent activity, acting on DNA / methylated histone binding / DNA-templated transcription initiation / helicase activity / transcription elongation by RNA polymerase II / double-strand break repair via homologous recombination / Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement / chromatin DNA binding / structural constituent of chromatin / nucleosome / nucleosome assembly / site of double-strand break / histone binding / transcription cis-regulatory region binding / chromatin remodeling / protein heterodimerization activity / chromatin binding / chromatin / regulation of transcription by RNA polymerase II / ATP hydrolysis activity / mitochondrion / DNA binding / ATP binding / nucleus Similarity search - Function | ||||||
| Biological species |  synthetic construct (others) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 10 Å | ||||||
 Authors Authors | Sundaramoorthy, R. / Owen-hughes, T. / Norman, D.G. / Hughes, A. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Elife / Year: 2018 Title: Structure of the chromatin remodelling enzyme Chd1 bound to a ubiquitinylated nucleosome. Authors: Ramasubramanian Sundaramoorthy / Amanda L Hughes / Hassane El-Mkami / David G Norman / Helder Ferreira / Tom Owen-Hughes / Abstract: ATP-dependent chromatin remodelling proteins represent a diverse family of proteins that share ATPase domains that are adapted to regulate protein-DNA interactions. Here, we present structures of the ...ATP-dependent chromatin remodelling proteins represent a diverse family of proteins that share ATPase domains that are adapted to regulate protein-DNA interactions. Here, we present structures of the Chd1 protein engaged with nucleosomes in the presence of the transition state mimic ADP-beryllium fluoride. The path of DNA strands through the ATPase domains indicates the presence of contacts conserved with single strand translocases and additional contacts with both strands that are unique to Snf2 related proteins. The structure provides connectivity between rearrangement of ATPase lobes to a closed, nucleotide bound state and the sensing of linker DNA. Two turns of linker DNA are prised off the surface of the histone octamer as a result of Chd1 binding, and both the histone H3 tail and ubiquitin conjugated to lysine 120 are re-orientated towards the unravelled DNA. This indicates how changes to nucleosome structure can alter the way in which histone epitopes are presented. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6g0l.cif.gz | 688.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6g0l.ent.gz | 539.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6g0l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6g0l_validation.pdf.gz | 1014.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6g0l_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | 6g0l_validation.xml.gz | 118 KB | Display | |
| Data in CIF | 6g0l_validation.cif.gz | 174 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g0/6g0lftp://data.pdbj.org/pub/pdb/validation_reports/g0/6g0l | HTTPS FTP |
-Related structure data
| Related structure data |  4336MC  4318C  6ftxC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
-Protein , 4 types, 8 molecules AEBFDHMW
| #1: Protein | Mass: 15407.075 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: Histone H3 sequence from X. Laevis. The N-terminal region from 1-39 is not resolved. Source: (gene. exp.)  #2: Protein | Mass: 11394.426 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: Histone H4 sequence from X. Laevis. The N-terminal region from 1-19 is not resolved. Source: (gene. exp.) #4: Protein | Mass: 13939.228 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #8: Protein | Mass: 168496.609 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P32657, Hydrolases; Acting on acid anhydrides; Acting on acid anhydrides to facilitate cellular and subcellular movement |
|---|
-Histone H2A type ... , 2 types, 2 molecules CG
| #3: Protein | Mass: 14193.578 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #5: Protein | Mass: 14093.436 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
-DNA chain , 2 types, 2 molecules IJ
| #6: DNA chain | Mass: 54088.469 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
|---|---|
| #7: DNA chain | Mass: 54885.938 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
-Non-polymers , 2 types, 4 molecules 
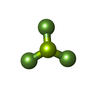
| #9: Chemical |  Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#10: Chemical |  Mass: 66.007 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: BeF3 Mass: 66.007 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: BeF3 |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.600 MDa / Experimental value: NO | ||||||||||||||||||||||||||||||
| Source (natural) |
| ||||||||||||||||||||||||||||||
| Source (recombinant) |
| ||||||||||||||||||||||||||||||
| Buffer solution | pH: 7.5 | ||||||||||||||||||||||||||||||
| Buffer component |
| ||||||||||||||||||||||||||||||
| Specimen | Conc.: 1 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES Details: Sample was gel filtration purified and it is monodisperse | ||||||||||||||||||||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 400 divisions/in. / Grid type: C-flat-1.2/1.3 4C | ||||||||||||||||||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK III / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 277 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 98591 X / Calibrated magnification: 98591 X / Nominal defocus max: 4000 nm / Nominal defocus min: 1500 nm / Calibrated defocus min: 1500 nm / Calibrated defocus max: 4000 nm / Cs: 2.7 mm / C2 aperture diameter: 70 µm / Alignment procedure: BASIC |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Temperature (max): 170 K / Temperature (min): 170 K |
| Image recording | Average exposure time: 2 sec. / Electron dose: 1.56 e/Å2 / Detector mode: INTEGRATING / Film or detector model: FEI FALCON III (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 1800 |
| Image scans | Sampling size: 14 µm / Width: 3870 / Height: 3870 / Movie frames/image: 32 / Used frames/image: 5-28 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 250000 | ||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 10 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 68000 / Algorithm: FOURIER SPACE / Num. of class averages: 1 / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | B value: 800 / Protocol: RIGID BODY FIT / Space: RECIPROCAL / Target criteria: Cross-correlation coefficient |
